
Abstract
Background
Lignocellulose biomass contains high amount of biotin and resulted in an excessive biotin condition for cellulosic glutamic acid accumulation by Corynebacterium glutamicum. Penicillin or ethambutol triggers cellulosic glutamic acid accumulation, but they are not suitable for practical use due to the fermentation instability and environmental concerns. Efficient glutamic acid production from lignocellulose feedstocks should be achieved without any chemical inductions.
Results
An industrial strain C. glutamicum S9114 was metabolically engineered to achieve efficient glutamic acid accumulation in biotin-excessive corn stover hydrolysate. Among the multiple metabolic engineering efforts, two pathway regulations effectively triggered the glutamic acid accumulation in lignocellulose hydrolysate. The C-terminal truncation of glutamate secretion channel MscCG (ΔC110) led to the successful glutamic acid secretion in corn stover hydrolysate without inductions. Then the α-oxoglutarate dehydrogenase complex (ODHC) activity was attenuated by regulating odhA RBS sequence, and glutamic acid accumulation was further elevated for more than fivefolds. The obtained C. glutamicum XW6 strain reached a record-high titer of 65.2 g/L with the overall yield of 0.63 g/g glucose using corn stover as the starting feedstock without any chemical induction.
Conclusions
Metabolic engineering method was successfully applied to achieve efficient glutamic acid in biotin-rich lignocellulose hydrolysate for the first time. This study demonstrated the high potential of glutamic acid production from lignocellulose feedstock.
Similar content being viewed by others

Background
Lignocellulose biomass is the most promising alternative feedstock for the production of glutamic acid that can be used as building block chemical [1]. However, no essential glutamic acid had been successfully produced from lignocellulose biomass until recently the rich biotin content in lignocellulose biomass was identified [2]. The rich biotin in lignocellulose biomass maintains stability during the biorefinery processing and creates an excessive condition for glutamic acid secretion [2]. Excessive biotin resulted in reduced carbon flux for glutamic acid biosynthesis [3] but strengthened cell wall barrier for glutamic acid secretion [2, 4, 5]. Therefore, no glutamic acid can be accumulated by Corynebacterium glutamicum cells in biotin-excessive lignocellulose hydrolysate [2]. The general approach to deal with high biotin concentration is the chemical inductions such as adding of β-lactam antibiotics, surfactants or ethambutol [6, 7]. Penicillin and ethambutol addition triggered glutamic acid accumulation in lignocellulose hydrolysate [2, 7]. However, more complicated cell growth and glutamic acid accumulation would be encountered by penicillin or ethambutol induction under the existence of excessive biotin and the typical inhibitors generated from pretreatment step, such as furfural, 5-hydroxymethylfurfural (HMF), and phenolic aldehydes [8]. Thus, only relatively low glutamic acid could be produced by these two triggering methods [2, 7]. Overuse of penicillin and ethambutol also causes environmental and health concerns. Therefore, metabolic engineering of C. glutamicum strain to achieve efficient glutamic acid production in lignocellulose hydrolysate is the best way for the production of cellulosic glutamic acid in industrial practice.
Among the genetic modifications to trigger glutamic acid accumulation under excessive biotin condition, knockout of some genes, such as dtsR [9], ltsA [10], and odhA [11], caused glutamic acid accumulation to a certain extent. However, these modifications often resulted in impaired or more susceptible cell growth. Modulating the expression of some lipid synthesis-related genes only led to limited glutamic acid secretion [12]. Therefore, they were not suitable for glutamic acid fermentation in harsh inhibitors containing lignocellulose hydrolysate system. Glutamic acid secretion channel modification is also a potential way to achieve active glutamic acid secretion [13, 14], but its effect on inhibitors containing lignocellulose hydrolysate environment was still unclear. The metabolic engineering of C. glutamicum on triggering efficient glutamic acid accumulation from lignocellulose feedstock should balance the glutamic acid secretion from high-biotin-containing hydrolysate and the negative impact of the inhibitors in the hydrolysates on cell growth. In this study, we tried to achieve the glutamic acid accumulation of C. glutamicum S9114 in corn stover hydrolysate by the activation of glutamic acid secretion and enhancement of glutamic acid synthesis pathway. Finally, a metabolically engineered C. glutamicum XW6 strain suitable for efficient cellulosic glutamic acid accumulation was obtained, and a record-high titer of glutamic acid was achieved using corn stover as feedstock. The engineered strain provides the first practically applicable basis for the production of commodity glutamic acid from lignocellulose biomass for building block chemical use.
Results
Triggering glutamic acid secretion by C-terminal truncation of glutamate exporter MscCG
Our previous study showed that the excessive biotin in corn stover completely blocked glutamic acid accumulation in the hydrolysate prepared from pretreated and detoxified corn stover [2]. In this study, we tried two metabolic modifications to facilitate glutamic acid secretion, either by restricting the biotin uptake to reduce the intracellular biotin content to a sub-optimal biotin level suitable for glutamate secretion [15], or by truncating the C-terminal amino acid residue to activate the activity of glutamate exporter MscCG [13, 14].
To reduce the biotin uptake to reach a sub-optimal level in the cells, we knocked out one of the biotin transporter BioYMN-encoding genes bioY by electro-transformation of pK18–ΔbioY into C. glutamicum S9114 cells following two-round positive selections. Compared to that of the parental C. glutamicum S9114, the resulting C. glutamicum ΔbioY mutant showed longer lag phase time of 12 h (Fig. 1a). The cell growth rate was decreased by about 50%, and the corresponding glucose consumption rate was reduced by about 36% compared to that of C. glutamicum S9114 (Fig. 1b). No glutamic acid accumulation was observed in high-sugar corn stover hydrolysate with excessive biotin (approximately 125 µg/L) (Fig. 1c) which was the same as that of C. glutamicum S9114. However, glutamic acid was accumulated when culturing the mutant in a less-biotin-containing corn stover hydrolysate (approximately 60 µg/L, but still excessive than the sub-optimal biotin of 2–5 µg/L) (Additional file 1: Figure S1). This result indicates that bioY knockout only blocked the biotin uptake partially, and glutamic acid can only be accumulated in low-biotin-containing lignocellulose system. However, high-biotin-containing system still drove sufficient biotin into the cells and failed in glutamic acid accumulation due to the existence of non-specific biotin uptake channel [16]. Therefore, the biotin-dependent glutamic acid secretion cannot be eliminated by bioY gene knockout.

Triggering glutamic acid accumulation by C-terminal truncation of MscCG and bioY knockout of C. glutamicum S9114. a Cell growth; b glucose consumption; c glutamic acid accumulation. Parental indicates the original C. glutamicum S9114 strain; ΔC110 indicates the strain with the MscCG C-terminal truncated; ΔbioY indicates the strain with bioY gene knocked out; ΔC110ΔbioY indicates the strain with the MscCG C-terminal truncated and bioY gene knocked out. Glutamic acid fermentations were carried out in 3-L fermentor (3BG-4, Baoxing Biotech Co., Shanghai, China) at 32 °C, 1.4 vvm of aeration and 600 rpm. No penicillin was added for induction. Mean values are presented with error bars representing the minimum and maximum values
For glutamic acid secretion, the Ncgl1221 gene-encoded glutamate secretion channel protein MscCG was responsible for modulating glutamate export, and its C-terminal region was accountable for the gating process of the channel [13, 14]. The C-terminal 110 amino acid residue truncation achieved glutamate secretion in biotin-excessive conditions without inductions in two different C. glutamicum strains [13, 14]. Thus, we tried to truncate its C-terminal residues to activate glutamic acid secretion here. The resulting C. glutamicum ΔC110 mutant showed similar cell growth rate with 16% reduced cell mass and similar glucose consumption rate, compared to that of C. glutamicum S9114 (Fig. 1a, b). However, glutamic acid accumulation was significantly elevated to 9.6 g/L within 32 h (Fig. 1c). This observed constitutive glutamic acid accumulation indicates the C-terminal truncation of MscCG is an effective method to achieve glutamic acid secretion without inductions in the harsh inhibitors and rich-biotin-containing corn stover hydrolysates. We further integrated the bioY knockout with MscCG C-terminal truncation by bioY knockout in the MscCG C-terminal truncated ΔC110 strain. The resulting double-knockout-mutant C. glutamicum ΔC110ΔbioY did not show further significant improvement in glutamic acid accumulation than the single-mutant C. glutamicum ΔC110, but the cell growth and glucose consumption rate were reduced to that of the C. glutamicum ΔbioY mutant (Fig. 1). Since bioY knockout was only effective in a relatively low-biotin concentration-containing corn stover hydrolysate, it was reasonable that it did not show any synergistic effect on glutamic acid secretion in the high-sugar corn stover hydrolysate with higher biotin concentration. However, impaired cell growth and glucose consumption would intensely affect glutamic acid productivity. Thus, bioY gene knockout in ΔC110 strain was also not suitable for efficient glutamic acid production and bioY gene knockout is not an ideal method to trigger glutamic acid accumulation in biotin-excessive corn stover hydrolysate. The effective MscCG C-terminal-truncated ΔC110 strain was used for the subsequent experiments.
Increasing glutamic acid accumulation by odhA attenuation
Decreasing α-oxoglutarate dehydrogenase complex (ODHC) activity is an essential approach to redirect α-oxoglutarate flux to glutamate synthesis instead of succinyl-CoA synthesis [6, 17]. Besides the disruption of odhA to completely remove the ODHC activity which blocks TCA cycle and severely inhibits cell growth [11], other methods were also used to decrease ODHC activity, such as changing RBS (ribosome-binding site) [18] or the translational start codon [19], using anti-odhA antisense RNA [20] and manipulating odhA inhibitory protein OdhI [21,22,23,24]. Among these methods, we considered changing RBS sequence as an easier but efficient way, because the enzyme activity can be decreased to certain levels by applying designed RBS sequences with different strength [18, 25]. Thus, the optimal ODHC activity that balances the carbon flux for the TCA cycle and glutamic acid synthesis can be reached. Based on the RBS calculator (https://www.denovodna.com/software/doLogin), the original RBS sequence of odhA was evaluated to be 50.38 a.u. (Table 1). Then three RBS sequences with strength of 20 a.u., 10 a.u. and 0.1 a.u. were designed and substituted the original RBS sequence in the genome of the MscCG C-terminal-truncated ΔC110 mutant based on the homologous sacB recombination system [26], and resulted in ΔC110odhARBS20, ΔC110odhARBS10 and ΔC110odhARBS0.1 mutant, respectively.
The following fermentation assay showed that RBS sequence substitution resulted in normal cell growth during the fermentation with about 30% reduced cell growth rate (Fig. 2a) compared to that of ΔC110 strain, indicating RBS optimization did not deprive the residual ODHC activity for cell growth. Glucose was completely consumed within 36 h compared to 32 h of ΔC110 strain (Fig. 2b). All of the RBS-substituted recombinants exhibited a significant increase in glutamic acid production compared to ΔC110 strain (Fig. 2c). All the three RBS-substituted recombinants produced more than 50 g/L glutamic acid, and the highest glutamic acid titer of 55.7 ± 0.1 g/L and yield of 53.5% within 36 h were achieved by ΔC110odhARBS0.1 strain, which was 5.8-fold higher than that of ΔC110 strain. Thus, decreasing ODHC activity by replacing the original odhA RBS sequence with designed RBS with the strength of 0.1 a.u. was suitable for cellulosic glutamic acid production. Higher glutamic acid titer and productivity (16.8% and 55.6% improvement, respectively), as well as more stable cell growth, were achieved compared to that of penicillin-triggered glutamic acid fermentation of C. glutamicum S9114. This engineered strain was named as C. glutamicum XW6 and used for the following experiments.

Improving glutamic acid accumulation by adjusting odhA RBS strength of ΔC110 strain. a Cell growth; b glucose consumption; c glutamic acid accumulation. Parental (with penicillin induction) indicates the penicillin-triggered glutamic acid fermentation by parental C. glutamicum S9114 strain. Penicillin induction method is described in “Methods”. ΔC110 (control) indicates the MscCG C-terminal-truncated strain; ΔC110odhARBS20, ΔC110odhARBS10 and ΔC110odhARBS0.1 indicate the ΔC110 strains with different odhA RBS strength of 20 a.u., 10 a.u. and 0.1 a.u. substituted, respectively. Glutamic acid fermentations of engineered recombinants were carried out in 3-L fermentor (3BG-4, Baoxing Biotech Co., Shanghai, China) at 32 °C, 1.4 vvm of aeration and 600 rpm. No penicillin was added for induction. Mean values were presented with error bars representing the minimum and maximum values
Overexpression of genes on glutamic acid synthesis pathway for accumulation enhancement
To further improve the glutamic acid production, the key genes involved in glutamic acid synthesis from phosphoenolpyruvate to glutamic acid were overexpressed in MscCG C-terminal-truncated and odhA gene RBS sequence-substituted C. glutamicum XW6 (ΔC110odhARBS0.1) strain. These selected genes include pyruvate carboxylase-encoding gene pyc and phosphoenolpyruvate carboxylase-encoding gene ppc [27]. Besides, the rate-limiting citrate synthase in the TCA cycle [28]-encoding gene gltA and the isocitrate dehydrogenase-encoding gene icd [29] were included. We also incorporated two glutamate dehydrogenase-encoding genes gdh1 and gdh2, as well as the modified glutamate exporter-encoding gene. Corresponding overexpression plasmids as well as the empty expression plasmid were electrotransformed into the C. glutamicum XW6 cells and then confirmed by colony PCR.
All of the gene overexpression recombinants grew well in the corn stover hydrolysate and produced more than 55 g/L of glutamic acid (Table 2). However, the overexpression of these genes did not result in significant improvement in glutamic acid titer and yield. Compared to that of the control, only 1 g/L higher glutamic acid titer of 60.5 g/L was achieved in the strain with overexpressed gltA. This higher glutamic acid titer may be due to the relatively higher initial glucose during the fermentation, and the glutamic acid productivity (1.26 ± 0.02 g L−1 h−1) and yield (54.0%) were both lower than that of control (1.36 ± 0.11 g L−1 h−1 and 54.5%, respectively). The modified glutamate exporter overexpression resulted in the same glutamic acid titer, but relatively higher glutamic acid yield (56.1% compared to 54.9%) and 9.6% higher productivity (1.49 ± 0.01 g L−1 h−1 compared to 1.36 ± 0.11 g L−1 h−1), compared to that of control. Therefore, overexpression of the modified glutamate exporter would enhance the glutamic acid secretion and benefit the glutamic acid production. We further tried to increase the modified glutamate exporter expression by integrating its encoding genes with H36 promoter to the genome of C. glutamicum at CGS9114_RS02700 gene locus which encoded a putative lactate dehydrogenase, and resulted in XW6-ΔRS02700::H36-ΔC110 strain. However, the glutamic acid fermentation of this strain (Additional file 1: Figure S2) did not show any improvement in glutamic acid production performance compared to that of XW6.
The other genes’ overexpression even led to lower glutamic acid titer and yield, and no significant improvement in glutamic acid productivity can be reached. Therefore, they had limited effects on improving glutamic acid accumulation here. The important role of pyruvate carboxylase and phosphoenolpyruvate carboxylase in oxaloacetate supply for glutamate production was extensively investigated before [27, 30, 31]. Phosphoenolpyruvate carboxylase often shows severe feedback inhibitions by many metabolic intermediates, while the overexpression of pyruvate carboxylase was more effective in biotin-limited condition because this enzyme requires biotin as co-factor [31]. These may be the reason why no improvement can be achieved by pyc and ppc overexpression. As for gltA and icd, their overexpression was supposed to channel more carbon flux to the supply of α-oxoglutarate. However, excessive accumulation of α-oxoglutarate due to the attenuated ODHC activity may offset the effects of gltA and icd overexpression. The overexpression of the two glutamate dehydrogenases still could not result in improved glutamate accumulation, and this was much the same with the previous report when glutamate dehydrogenase expression level was higher than 13.5-fold in an odhA-disrupted mutant [11]. Since H36 was a strong promoter used for high-level gene expression in Corynebacterium glutamicum [32], these genes’ overexpression under the control of the strong H36 promoter may not be suitable, and the metabolic burden caused by introducing these expression plasmids should also be taken into consideration. Further investigation should be addressed to show how these genes affect the glutamic acid accumulation in C. glutamicum XW6 strain. Just the overexpression of these genes was not effective for improving glutamic acid production in this glutamate-overproducing bacterium. The primary by-product of lactic acid was not formed during all the fermentations of XW6 strain and its derivatives. Therefore, this two-step metabolic-engineered strain (XW6) obtained by activating glutamic acid secretion and attenuating of odhA was used for high-titer glutamic acid production in the biotin-excessive corn stover hydrolysate.
Evaluation of glutamic acid fermentation from corn stover hydrolysate
The cellulosic glutamic acid fermentation was conducted for the evaluation of the engineered strain in corn stover hydrolysate. Cells grew well during the fermentation process, cell mass reached a peak at 30 h and maintained at a certain level after the addition of fresh corn stover hydrolysate (Fig. 3). With continuous glucose consumption, glutamic acid continued to accumulate and reached the maximum glutamic acid titer of 65.2 ± 1.4 g/L within 48 h (Fig. 3). Finally, 149.0 ± 1.6 g glucose was consumed and 93.9 ± 2.0 g glutamic acid was produced. The yield based on consumed glucose reached 63.0 ± 0.7%. The glutamic acid titer is record high for glutamic acid production using lignocellulose biomass.

Cellulosic glutamic acid fermentation by the engineered C. glutamicum XW6 strain in corn stover hydrolysate. Glutamic acid fermentation was carried out in 3-L fermentor (3BG-4, Baoxing Biotech Co., Shanghai, China) at 32 °C, 1.4 vvm of aeration and 600 rpm. No penicillin was added for induction. The fermentation was initiated by inoculating the seed culture in the 800-mL-corn stover hydrolysate-containing fermentor, and then 125 mL, 150 mL and 175 mL of fresh corn stover hydrolysate was fed into the fermentor at 30 h, 36 h and 40 h, respectively. Mean values are presented with error bars representing the minimum and maximum values
Discussion
For metabolic engineering of C. glutamicum to achieve glutamic acid accumulation under biotin-excessive condition, many reported modifications such as dtsR [9], ltsA [10], odhA [11] knockout often resulted in impaired or more susceptible cell growth. Considering the excessive biotin and harsh inhibitors in lignocellulose hydrolysate, they were not suitable for triggering glutamic acid production in lignocellulose system. Metabolic engineering methods should balance the glutamic acid accumulation and impacts on cell growth. In this study, we tried many methods for metabolic engineering of C. glutamicum S9114 for glutamic acid production in biotin-excessive corn stover hydrolysate. Among the methods we carefully screened and tested (Fig. 4), glutamic acid secretion channel modification (C-terminal truncation of MscCG) and odhA attenuation by RBS optimization were the most effective ways. These two metabolic modifications successfully triggered glutamic acid accumulation in biotin and inhibitors containing lignocellulose hydrolysate without any penicillin usage. We also tried bioY gene knockout and glutamic acid synthesis enhancement, but only slight improvement of glutamic acid accumulation was observed (Fig. 4). Thus, the MscCG C-terminal-truncated and the odhA gene RBS sequence-substituted strain XW6 was selected for cellulosic glutamic acid fermentation using corn stover as feedstock after dry acid pretreatment, biodetoxification and enzymatic hydrolysis, and a record-high glutamic acid titer (65.2 g/L) was obtained. Although this glutamic acid titer was still relatively low compared to that of the starch-based glutamic acid fermentation (over 120 g/L) [2], the potential of the engineered strain for cellulosic glutamic acid production using biotin-rich lignocellulose feedstock was fully demonstrated. Future metabolic engineering to co-utilize the lignocellulose-derived pentose sugars and oligomer sugars in lignocellulose hydrolysate such as the xylose, arabinose and cellobiose would further promote the cellulosic glutamic acid production towards industrial application.

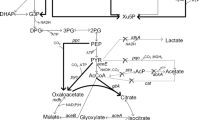
Illustration of Corynebacterium glutamicum engineering methods for efficient cellulosic glutamic acid production. The left chart indicates the overall metabolic pathway modifications of C. glutamicum; the right chart indicates the elevations of glutamic acid accumulation corresponding to each step of metabolic engineering modification. PEP phosphoenolpyruvate, Pyr pyruvate, AcCoA acetyl-CoA, Cit citrate, Icit isocitrate, 2-KG α-oxoglutarate; Suc succinate, Fum fumarate, Mal malate, OAA oxaloacetate, Gly glyoxylate, AccBC acetyl-CoA carboxylase α-subunit, AccD1 acetyl-CoA carboxylase β-subunit. All the fermentations were carried out in 3-L fermentor (3BG-4, Baoxing Biotech Co., Shanghai, China) at 32 °C, 1.4 vvm of aeration and 600 rpm. No penicillin was added for induction. Mean values are presented with error bars representing the minimum and maximum values
Conclusions
Improved non-chemical-induced glutamic acid production in biotin-excessive lignocellulose hydrolysate was successfully achieved using a metabolic-engineered C. glutamicum strain, compared to the penicillin-triggered glutamic acid fermentation by the parental strain. A record high titer of 65.2 ± 1.4 g/L of glutamic acid was produced from corn stover hydrolysate. The biorefinery application of this engineered strain for cellulosic glutamic acid production was fully demonstrated.
Methods
Strains, media and culture conditions
Strains and plasmids used in this study are listed in Table 3. Escherichia coli DH5α was cultured on Luria–Bertani (LB) medium with 50 μg/mL of kanamycin addition if needed. C. glutamicum S9114 was purchased from the Shanghai Industrial Institute of Microorganism (SIIM, http://www.gsy-siim.com/), Shanghai, China, with the storage code of B460. The culture medium was CM2B and contained 10 g/L of peptone, 10 g/L of yeast extract, 10 g/L of NaCl and 10 μg/L of biotin (pH 7.0). The culture condition was 30 °C, 200 rpm, and 50 μg/mL of kanamycin was added if needed. Amorphotheca resinae ZN1 was used for biodetoxification, and it was cultured on PDA medium containing 200 g of potato juice, 20 g of glucose and 17 g of agar per liter at 28 °C [8].
Enzyme and reagents
Cellulase Cellic CTec2 was purchased from Novozymes (China), Beijing, China. The filter paper activity was determined to be 203.2 FPU/mL according to NREL protocol LAP-006 [33]; cellobiase activity was determined to be 4900 CBU/mL according to the method reported previously [34]. Total protein concentration was 87.3 mg/mL based on the Bradford method [35]. DNA polymerase and T4 ligase were purchased from Takara, Otsu, Japan. Restriction endonucleases were purchased from Thermo Scientific, Wilmington, DE, USA. Seamless cloning kit HB-infusionTM was purchased from Hanheng Biotech Co., Nanjing, China. Penicillin G with the titer of 1650 U/mg was purchased from New Probe Biochem Co., Beijing, China. Other general chemicals used in this study were of analytical grade and purchased from local suppliers.
Plasmid and recombinant construction
Plasmids and recombinant strains constructed in this study are listed in Table 3 and the primers used are listed in Additional file 1: Table S1. The upstream and downstream fragments of bioY gene were amplified from the genome of C. glutamicum S9114 using the primer pairs of bioY-up-F/R and bioY-down-F/R, and subsequently inserted into XbaI/PstI and PstI/HindIII of pK18mobsacB, respectively, resulting in pK18–ΔbioY. The upstream and downstream fragments of the corresponding C-terminal 110-amino acid-encoding sequence were amplified from the genome by the primer pairs of ΔC110-up-F/R and ΔC110-down-F/R, and then these fragments were overlapped together by PCR. The fused PCR product was then inserted into BamHI/XbaI of pK18mobsacB to generate pK18–ΔC110. For the construction of RBS substitution plasmids of odhA gene, three RBS sequences with different strength of 0.1 a.u., 10 a.u. and 20 a.u. were first designed by the RBS calculator (https://www.denovodna.com/software/doLogin). Primer pairs of odhA-up-F/odhA-RBS0.1-up-R and odhA-RBS0.1-down-F/odhA-down-R were used to amplify the upstream and downstream of the original RBS sequence from the genome, and assembled together with EcoRI/BamHI-linearized pK18mobsacB fragment by seamless cloning kit, resulting pK18–odhARBS0.1. The same method was applied to construct pK18–odhARBS10 and pK18–odhARBS20 using corresponding primer pairs. For the construction of pK18–ΔRS02700::H36ΔC110, H36 promoter and MscCGΔC110-encoding sequence were amplified from the pH36mob plasmid and C. glutamicum S9114 genome, respectively, using primer pairs of H36-F/R and ΔC110-overlap-F/R. Then the up- and downstream of CGS9114_RS02700 were amplified from the genome using primer pairs of RS02700-up-F/R and RS02700-down-F/R. After fused together by overlap PCR using primers of RS02700-up-F/RS02700-down-R, it was assembled together with XbaI/HindIII-linearized pK18mobsacB, resulting pK18–ΔRS02700::H36ΔC110.
The pH36mob plasmid for gene expression used in this study was first constructed by replacing the Trc promoter of pTRCmob with a strong synthetic promoter H36 [32] which was carried out by Shanghai Generay Biotech Co., Ltd, Shanghai, China. The fragment encoding C-terminal-truncated MscCG (MscCGΔC110) was amplified from C. glutamicum S9114 genome using primers ΔC110-F/R, and then it was inserted into XbaI/SalI restriction site of pH36mob to generate pH36–ΔC110. The fragment of NADPH-dependent glutamate dehydrogenase-encoding gene gdh1 and another glutamate dehydrogenase-encoding gene gdh2 was amplified from the genome using primers gah1-F/R and gdh2-F/R and then inserted to XbaI/SalI and XbaI/PstI of pH36mob, respectively, resulting in pH36-gdh1 and pH36-gdh2. The citrate synthase-encoding gene gltA and isocitrate dehydrogenase-encoding gene icd were amplified from the genome using primers gltA-F/R and icd-F/R, respectively. Then these two fragments were inserted into EcoRI/XbaI of pH36mob to generate pH36–gltA and pH36–icd. The pH36-pyc plasmid was constructed by ligating the pyruvate carboxylase-encoding gene pyc fragment amplified using primers pyc-F/R and the PCR using pH36-vector-F/R-linearized pH36mob together by the seamless cloning kit. Phosphoenolpyruvate carboxylase-encoding gene ppc was amplified from the genome using primers of ppc-F/R, and inserted into XbaI/SalI site of pH36mob, resulting in pH36–ppc. All the plasmids constructed were sequenced right before use.
Plasmids were transformed into C. glutamicum cells by electroporation [36]. The gene disruption, integration and RBS sequence substitution in the genome were based on homologous sacB recombination system [26]. The correct mutants were isolated through two rounds of homologous recombination and confirmed by colony PCR and sequence analysis. Single colonies with kanamycin resistance were picked up and then confirmed by colony PCR using primers pH36-F and corresponding reverse primers of different genes.
Lignocellulose feedstock and biorefinery processing
Corn stover was harvested in fall 2016, Tongliao, Inner Mongolia, China. The raw biomass was air dried and milled, and then the composition was determined to be 33.0% cellulose, 26.9% hemicellulose, 20.8% lignin and 6.3% ash according to National Renewable Energy Laboratory (NREL) protocols [37, 38]. The dry acid pretreatment was carried out according to our previous procedure [39, 40], and 3.8% (w/w, based on dry material matter) H2SO4 was used for pretreatment. The pretreated corn stover contained 40.1 mg of glucose, 132.7 mg of xylose, 5.1 mg of furfural, 9.7 mg of 5-hydroxymethylfurfural (HMF), and 19.2 mg of acetic acid per gram of dry corn stover (dry matter, DM). The pretreated corn stover was then biodetoxified according to our previous procedure [8, 41]. Corn stover hydrolysate was prepared by hydrolyzing the pretreated and biodetoxified corn stover material at a solid content of 30% (w/w) dry corn stover solids at a cellulase usage of 10 mg proteins per gram for 48 h at 50 °C. The hydrolysate slurries were centrifuged at 16,125×g for 10 min to remove the solid residues and obtain clear supernatant hydrolysate. Then the hydrolysate was autoclaved by a high-pressure steam sterilizer (TOMY XS-700, Tomy Seiko, Co., Tokyo, Japan) at 115 °C for 20 min and filtrated by sterilized filter paper before use. The corn stover hydrolysate was determined to contain 131.6 ± 0.2 g/L glucose, 18.8 ± 0.1 g/L xylose, 2.3 ± 0.0 g/L acetic acid, 0.04 ± 0.01 g/L furfural and 0.02 ± 0.01 g/L HMF.
Glutamic acid fermentation
Corynebacterium glutamicum cells were cultured on CM2B agar at 30 °C for 24–36 h, and then a single colony was picked up for preparing the seed culture as described previously [2]. The batch fermentation was performed in a 3-L fermentor (3BG-4, Baoxing Biotech Co., Shanghai, China) at 32 °C, 1.4 vvm of aeration and 600 rpm. The seed culture was inoculated in 800 mL of corn stover hydrolysate at 10% (v/v) inoculum ratio. The pH was maintained at 7.2 by automatic addition of 25% (w/v) ammonium hydroxide solution. If needed, 50 μg/mL of kanamycin was added to the hydrolysate for maintaining the plasmid. The penicillin-induced glutamic acid fermentation by C. glutamicum S9114 followed the same procedure as reported before [2]. All fermentations were carried out in duplicate.
Analytical methods
Glucose, glutamic acid and lactic acid were analyzed using the SBA-90 biosensor (Biology Institute, Shandong Academy of Sciences, Jinan, Shandong, China). Xylose acetic acid, furfural, and HMF were analyzed on HPLC (LC-20AD, Shimadzu, Kyoto, Japan) equipped with a Bio-rad Aminex HPX-87H column (Bio-rad, USA) and RID-10A detector (Shimadzu, Kyoto, Japan) according to the method reported before [41]. Phenolic compounds were analyzed on HPLC (UV/Vis detector SPD-20A, at 270 nm, Shimadzu, Kyoto, Japan) with a YMC-Pack ODS-A column (YMC Co., Kyoto, Japan) at 35 °C as mentioned before [41]. Biotin concentration in corn stover hydrolysate was determined as reported previously [2]. Cell growth was indicated by optical density at 600 nm (OD600) by the UV–visible spectrophotometer BIOMATE 3S (Thermo, Waltham, MA, USA).
Glutamic acid yield calculation
The glutamic acid yield based on the consumed glucose was calculated by the following equation:
where [GMA] and [GMA]0 indicate the final and initial concentrations of glutamic acid, respectively, [G]0 and [G] indicate the initial and final concentrations of glucose, respectively, V0 and V indicate the final and initial volumes of the fermentation broth, respectively. The volume change caused by the addition of ammonium hydroxide solution for pH maintenance was ignored in batch fermentation.
Abbreviations
- CSH:
-
corn stover hydrolysate
- HMF:
-
5-hydroxymethylfurfural
- ODHC:
-
α-ketoglutarate dehydrogenase complex
- MscCG:
-
mechanosensitive channel
- DM:
-
dry matter
- NREL:
-
National Renewable Energy Laboratory
- SIIM:
-
Shanghai Industrial Institute of Microorganism
- CGMCC:
-
China General Microorganism Collection Center
- PDA:
-
potato dextrose agar
- FPU:
-
filter paper unit
- CBU:
-
cellobiase unit
- OD600 :
-
optical density at wavelength (λ) 600 nm
- PEP:
-
phosphoenolpyruvate
- Pyr:
-
pyruvate
- AcCoA:
-
acetyl-CoA
- Cit:
-
citrate
- Icit:
-
isocitrate
- 2-KG:
-
α-oxoglutarate
- Suc:
-
succinate
- Fum:
-
fumarate
- Mal:
-
malate
- OAA:
-
oxaloacetate
- Gly:
-
glyoxylate
- AccBC:
-
acetyl-CoA carboxylase α-subunit
- AccD1:
-
acetyl-CoA carboxylase β-subunit
References
Jonsson LJ, Alriksson B, Nilvebrant NO. Bioconversion of lignocellulose: inhibitors and detoxification. Biotechnol Biofuels. 2013;6:16.
Wen J, Xiao Y, Liu T, Gao Q, Bao J. Rich biotin content in lignocellulose biomass plays the key role in determining cellulosic glutamic acid accumulation by Corynebacterium glutamicum. Biotechnol Biofuels. 2018;11:132.
Kimura E. Triggering mechanism of L-glutamate overproduction by DtsR1 in coryneform bacteria. J Biosci Bioeng. 2002;94:545–51.
Kimura E, Yagoshi C, Kawahara Y, Ohsumi T, Nakamatsu T, Tokuda H. Glutamate Overproduction in Corynebacterium glutamicum triggered by a decrease in the level of a complex comprising DtsR and a biotin-containing subunit. Biosci Biotechnol Biochem. 1999;63:1274–8.
Eggeling L, Krumbach K, Sahm H. l-Glutamate efflux with Corynebacterium glutamicum: why is penicillin treatment or tween addition doing the same? J Mol Microb Biotechnol. 2001;3:67–8.
Kawahara Y, Takahashi-Fuke K, Shimizu E, Nakamatsu T, Nakamori S. Relationship between the glutamate production and the activity of 2-oxoglutarate dehydrogenase in Brevibacterium lactofermentum. Biosci Biotechnol Biochem. 1997;61:1109–12.
Gopinath V, Meiswinkel TM, Wendisch VF, Nampoothiri KM. Amino acid production from rice straw and wheat bran hydrolysates by recombinant pentose-utilizing Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2011;92:985–96.
Zhang J, Zhu Z, Wang X, Wang N, Wang W, Bao J. Biodetoxification of toxins generated from lignocellulose pretreatment using a newly isolated fungus, Amorphotheca resinae ZN1, and the consequent ethanol fermentation. Biotechnol Biofuels. 2010;3:26.
Kimura E, Abe C, Kawahara Y, Nakamatsu T, Tokuda H. A dtsR gene-disrupted mutant of Brevibacterium lactofermentum requires fatty acids for growth and efficiently produces l-glutamate in the presence of an excess of biotin. Biochem Biophys Res Commun. 1997;234:157–61.
Hirasawa T, Wachi M, Nagai K. A mutation in the Corynebacterium glutamicum ltsA gene causes susceptibility to lysozyme, temperature-sensitive growth, and l-glutamate production. J Bacteriol. 2000;182:2696–701.
Asakura Y, Kimura E, Usuda Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T. Altered metabolic flux due to deletion of odhA causes l-glutamate overproduction in Corynebacterium glutamicum. Appl Environ Microbiol. 2007;73:1308–19.
Nampoothiri K, Hoischen C, Bathe B, Möckel B, Pfefferle W, Krumbach K, Sahm H, Eggeling L. Expression of genes of lipid synthesis and altered lipid composition modulates l-glutamate efflux of Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2002;58:89–96.
Becker M, Borngen K, Nomura T, Battle AR, Marin K, Martinac B, Kramer R. Glutamate efflux mediated by Corynebacterium glutamicum MscCG, Escherichia coli MscS, and their derivatives. Biochim Biophys Acta. 2013;1828:1230–40.
Yamashita C, Hashimoto K, Kumagai K, Maeda T, Takada A, Yabe I, Kawasaki H, Wachi M. l-Glutamate secretion by the N-terminal domain of the Corynebacterium glutamicum NCgl1221 mechanosensitive channel. Biosci Biotechnol Biochem. 2013;77:1008–13.
Takinami K, Yamada Y, Okada H. Biochemical effects of fatty acid and its derivatives on l-glutamic acid fermentation: part IV biotin content of growing cells of Brevibacterium lactofermentum. Agric Biol Chem. 1966;30:674–82.
Hebbeln P, Rodionov DA, Alfandega A, Eitinger T. Biotin uptake in prokaryotes by solute transporters with an optional ATP-binding cassette-containing module. Proc Natl Acad Sci USA. 2007;104:2909–14.
Hasegawa T, Hashimoto K, Kawasaki H, Nakamatsu T. Changes in enzyme activities at the pyruvate node in glutamate-overproducing Corynebacterium glutamicum. J Biosci Bioeng. 2008;105:12–9.
Zhang B, Yu M, Zhou Y, Li Y, Ye BC. Systematic pathway engineering of Corynebacterium glutamicum S9114 for l-ornithine production. Microb Cell Fact. 2017;16:158.
Nguyen AQ, Schneider J, Reddy GK, Wendisch VF. Fermentative production of the diamine putrescine: system metabolic engineering of Corynebacterium glutamicum. Metabolites. 2015;5:211–31.
Kim J, Hirasawa T, Sato Y, Nagahisa K, Furusawa C, Shimizu H. Effect of odhA overexpression and odhA antisense RNA expression on Tween-40-triggered glutamate production by Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2009;81:1097–106.
Kim J, Hirasawa T, Saito M, Furusawa C, Shimizu H. Investigation of phosphorylation status of OdhI protein during penicillin- and Tween 40-triggered glutamate overproduction by Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2011;91:143–51.
Komine-Abe A, Nagano-Shoji M, Kubo S, Kawasaki H, Yoshida M, Nishiyama M, Kosono S. Effect of lysine succinylation on the regulation of 2-oxoglutarate dehydrogenase inhibitor, OdhI, involved in glutamate production in Corynebacterium glutamicum. Biosci Biotechnol Biochem. 2017;81:2130–8.
Schultz C, Niebisch A, Gebel L, Bott M. Glutamate production by Corynebacterium glutamicum: dependence on the oxoglutarate dehydrogenase inhibitor protein OdhI and protein kinase PknG. Appl Microbiol Biotechnol. 2007;76:691–700.
Zhang C, Li Y, Ma J, Liu Y, He J, Li Y, Zhu F, Meng J, Zhan J, Li Z, et al. High production of 4-hydroxyisoleucine in Corynebacterium glutamicum by multistep metabolic engineering. Metab Eng. 2018;49:287–98.
Espah Borujeni A, Salis HM. Translation initiation is controlled by RNA folding kinetics via a ribosome drafting mechanism. J Am Chem Soc. 2016;138:7016–23.
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145:69–73.
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Mockel B, Sahm H, Eikmanns BJ. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microb Biotechnol. 2001;3:295–300.
Zhu N, Xia H, Wang Z, Zhao X, Chen T. Engineering of acetate recycling and citrate synthase to improve aerobic succinate production in Corynebacterium glutamicum. PLoS ONE. 2013;8:e60659.
Eikmanns BJ, Rittmann D, Sahm H. Cloning, sequence analysis, expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J Bacteriol. 1995;177:774–82.
Wada M, Sawada K, Ogura K, Shimono Y, Hagiwara T, Sugimoto M, Onuki A, Yokota A. Effects of phosphoenolpyruvate carboxylase desensitization on glutamic acid production in Corynebacterium glutamicum ATCC 13032. J Biosci Bioeng. 2016;121:172–7.
Guo X, Wang J, Xie X, Xu Q, Zhang C, Chen N. Enhancing the supply of oxaloacetate for l-glutamate production by pyc overexpression in different Corynebacterium glutamicum. Biotechnol Lett. 2013;35:943–50.
Yim SS, An SJ, Kang M, Lee J, Jeong KJ. Isolation of fully synthetic promoters for high-level gene expression in Corynebacterium glutamicum. Biotechnol Bioeng. 2013;110:2959–69.
Adney B, Baker J. Measurement of cellulase activities. NREL/TP-510-42628 Laboratory analytical procedure (LAP). National Renewable Energy Laboratory: Golden; 1996.
Ghose TK. Measurement of cellulase activities. Pure Appl Chem. 1987;59:257–68.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
van der Rest ME, Lange C, Molenaar D. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol. 1999;52:541–5.
Sluiter A, Hames B, R. Ruiz CS, Sluiter J, Templeton D, Crocker D. Determination of structural carbohydrates and lignin in biomass. Golden: NREL; 2012. NREL/TP-510-42618.
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D. Determination of sugars, byproducts, and degradation products in liquid fraction process samples. Golden: NREL; 2008. NREL/TP-510-42623.
He Y, Zhang J, Bao J. Dry dilute acid pretreatment by co-currently feeding of corn stover feedstock and dilute acid solution without impregnation. Bioresour Technol. 2014;158:360–4.
Zhang J, Wang X, Chu D, He Y, Bao J. Dry pretreatment of lignocellulose with extremely low steam and water usage for bioethanol production. Bioresour Technol. 2011;102:4480–8.
He Y, Zhang J, Bao J. Acceleration of biodetoxification on dilute acid pretreated lignocellulose feedstock by aeration and the consequent ethanol fermentation evaluation. Biotechnol Biofuels. 2016;9:19.
Wang Y, Cao G, Xu D, Fan L, Wu X, Ni X, Zhao S, Zheng P, Sun J, Ma Y. A novel Corynebacterium glutamicum l-glutamate exporter. Appl Environ Microbiol. 2018;84:e02691-17.
Authors’ contributions
JB conceived the study. JBW and JB designed the experiment and drafted the manuscript. JBW carried out all the experiments. Both authors read and approved the final manuscript.
Acknowledgements
Special thanks are addressed to Prof. Jibin Sun of Tianjin Institute of Industrial Biotechnology, Chinese Academy of Science, for kindly providing plasmids pTRCmob and pK18mobsacB.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This research was partially supported by the Natural Science Foundation of China (31961133006) and the National Key Research and Development Program of China (2017YFB0309302).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Primers used in this study. Figure S1. Effect of bioY gene knockout on glutamic acid accumulation in 15% (w/w) CSH. Figure S2. Glutamic acid fermentation by strain XW6-ΔRS02700::H36-ΔC110 in corn stover hydrolysate.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wen, J., Bao, J. Engineering Corynebacterium glutamicum triggers glutamic acid accumulation in biotin-rich corn stover hydrolysate. Biotechnol Biofuels 12, 86 (2019). https://doi.org/10.1186/s13068-019-1428-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-019-1428-5