
Abstract
Background
2,5-Furandicarboxylic acid is a renewable building block for the production of polyfurandicarboxylates, which are biodegradable polyesters expected to substitute their classical counterparts derived from fossil resources. It may be produced from bio-based 5-hydroxymethylfurfural or 5-methoxymethylfurfural, both obtained by the acidic dehydration of biomass-derived fructose. 5-Methoxymethylfurfural, which is produced in the presence of methanol, generates less by-products and exhibits better storage stability than 5-hydroxymethylfurfural being, therefore, the industrial substrate of choice.
Results
In this work, an enzymatic cascade involving three fungal oxidoreductases has been developed for the production of 2,5-furandicarboxylic acid from 5-methoxymethylfurfural. Aryl-alcohol oxidase and unspecific peroxygenase act on 5-methoxymethylfurfural and its partially oxidized derivatives yielding 2,5-furandicarboxylic acid, as well as methanol as a by-product. Methanol oxidase takes advantage of the methanol released for in situ producing H2O2 that, along with that produced by aryl-alcohol oxidase, fuels the peroxygenase reactions. In this way, the enzymatic cascade proceeds independently, with the only input of atmospheric O2, to attain a 70% conversion of initial 5-methoxymethylfurfural. The addition of some exogenous methanol to the reaction further improves the yield to attain an almost complete conversion of 5-methoxymethylfurfural into 2,5-furandicarboxylic acid.
Conclusions
The synergistic action of aryl-alcohol oxidase and unspecific peroxygenase in the presence of 5-methoxymethylfurfural and O2 is sufficient for the production of 2,5-furandicarboxylic acid. The addition of methanol oxidase to the enzymatic cascade increases the 2,5-furandicarboxylic acid yields by oxidizing a reaction by-product to fuel the peroxygenase reactions.
Similar content being viewed by others


Background
Fossil resources are finite and the need for substituting petroleum-based materials with renewable materials is increasing in recent years [1]. 2,5-Furandicarboxylic acid (FDCA) is nowadays regarded as a promising precursor for the production of renewable and biodegradable bioplastics. Polyester formed by the condensation of this building block with ethylene glycol, known as poly(ethylene-2,5-furandicarboxylate) (PEF), is expected to substitute for other polyesters produced from fossil fuels, thanks to their renewable origin and their mechanical and gas barrier properties, which are even better than those of conventional poly(ethylene terephthalate) (PET) [2, 3]. Therefore, it is expected that PEF will be able to compete with PET not only in economic but also in environmental terms since its production lowers the balance of green-house gases emissions [4]. The first report on PEF enzymatic hydrolysis, which permits the recycling of its monomers, has been brought to light recently [5].
FDCA can be obtained from precursors that are formed upon the acidic dehydration of fructose, directly obtained from plants (as monosaccharide, in sucrose disaccharide and in inulin-type polymers) or by isomerization of glucose from hydrolysis of disaccharides (e.g., sucrose) or polysaccharides (e.g., in lignocellulosic materials). These precursors are mainly 5-hydroxymethylfurfural (HMF) and more recently 5-methoxymethylfurfural (MMF). The latter is obtained when fructose is dehydrated in the presence of methanol or by HMF etherification [6,7,8]. MMF is more stable upon storage than HMF, and fructose dehydration in methanol yields less side-products than when it takes place in water for HMF production. Successful attempts have been made to obtain polyesters from MMF and its derivatives [9], and a joint venture between BASF and Avantium, Synvina (www.synvina.com), has been created for sustainable industrial production of PEF from stable MMF.
In the above context, several patents [10,11,12] present methods for the production of FDCA from MMF, but all of them use oxidation catalysts such as bromide, cobalt, or manganese, along with other metals. Moreover, they describe processes that take place at high temperatures (in the range of 100–220 °C) and pressures (3–15 bar). The advantage of enzymes, which work under mild conditions (in aqueous solution, at room temperature and under atmospheric pressure), for the production of FDCA has gained momentum and several reports on the enzymatic oxidation of HMF to FDCA are available [13,14,15]. Particularly, the use of the natural portfolio of oxidases and peroxygenases in synthetic chemistry is very timely. While the former can perform selective oxidations producing H2O2 from atmospheric O2, the latter can use the released H2O2 to complete the full oxidation of complex molecules like in the whole conversion of HMF to FDCA which comprises three sequential oxidation steps [13].
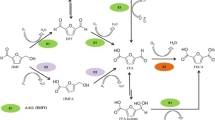
MMF conversion into FDCA can involve three or four oxidation steps depending on whether the ether breakdown leaves an alcohol or a carbonyl function in the furfural molecule (Fig. 1 scheme, pathways 1/3–5 or 1/2/5, respectively). In the present study, a new self-sustained enzymatic cascade was developed for the production of FDCA from MMF—combining aryl-alcohol oxidase (AAO) [16], unspecific peroxygenase (UPO) [17], and methanol oxidase (MOX) [18]—identified the intermediate products by gas chromatography-mass spectrometry (GC–MS), estimated the conversion yields and established the oxidation pathway.

Scheme of the possible pathways for the oxidation of MMF into FDCA. MMF, 5-methoxymethylfurfural; MMFA, 5-methoxymethylfurancarboxylic acid; HMFCA, 5-hydroxymethyl-2-furancarboxylic acid; FFCA, 5-formylfurancarboxylic acid; and FDCA, 2,5-furandicarboxylic acid
Results
Hydration of the carbonyl group in MMF
Comparison of 1H-NMR spectra in deuterated water and deuterated dimethylsulfoxide (DMSO-d6) allows detection of the aldehyde and the geminal diol signals to measure the degree of hydration at equilibrium. The MMF spectrum in DMSO-d6 showed six signals assigned to the aldehyde (9.6 ppm), the furanic ring (7.5 and 6.7 ppm), methylene ether (4.5 ppm), methyl (3.3 ppm), and residual DMSO (2.5 ppm) protons. On the contrary, the spectrum in sodium phosphate (pD 7.0) gave 8 signals assigned to the aldehyde proton (9.6 ppm) and its shifted counterpart hemiacetalic hydrated form (small signal at 6.8 ppm), the two ring protons (7.7 and 6.9 ppm) and their shifted counterparts (small signal at 6.5 ppm), the methylene ether (4.7 ppm) and methyl (3.5 ppm) protons, as well as the water protons (4.9 ppm). Integration of the aldehyde signal and its small shifted counterpart points towards a degree of MMF hydration ≤ 10%.
MMF oxidation by AAO
AAO may oxidize aldehydes to acids if their carbonyl groups are partially hydrated to gem-diols [19]. To test the ability of AAO from Pleurotus eryngii to oxidize the gem-diol form of MMF, the compound was incubated with the enzyme and the reaction was analyzed by GC–MS. The reaction was completed after 15 h, using a substrate/enzyme ratio of 300 under the conditions described above (Fig. 2a). 5-(Methoxymethyl)-2-furancarboxylic acid (MMFA) accumulated over time and additional products were not detected, confirming that AAO does not show any activity on the resulting molecule. The above results show that the small hydration degree shown by NMR was enough for AAO oxidation of the MMF molecule to MMFA (step 1 in Fig. 1 scheme).

Time course of the reaction of MMF (1.5 mM) with: a AAO; b AAO, UPO and H2O2; c AAO, UPO and MOX; and d AAO, UPO, MOX and methanol (at 28 °C in 100 mM phosphate, pH 7). Enzyme concentrations were 5 µM (AAO and UPO) and 1 µM (MOX), while H2O2 and methanol final concentrations (in b and d, respectively) were 1.5 mM (added after 24, 48 and 72 h) and 1 mM (added after 72 and 96 h reaction). Dashed lines in a and b represent the AAO and UPO residual activities as a function of time, respectively. Compounds were identified and quantified by GC–MS, using the estimated response factors
The kinetic constants for the AAO oxidation of MMF and related furfurals were indirectly measured as H2O2 release, by coupling the reaction of horseradish peroxidase (HRP) and a reagent that gives a colored product when peroxide is available (Table 1). Comparison with the kinetic constants estimated for related furfurals shows that the methoxyl moiety in MMF decreases the enzyme affinity (Km is increased) and lowers reactivity (kcat is slightly reduced). Both effects together result in much lower AAO catalytic efficiency for MMF than for the other two furfurals assayed.
Residual activity estimations showed that AAO was active during the whole time of the reaction (dashed line in Fig. 2a) displaying a half-life of around 6 h that guaranteed the full conversion of MMF into MMFA. Catalytic performance parameters of AAO, including residual activity together with turnover number (TON), turnover frequency (TOF), and total turnover number (TTN) values under the described conditions, are provided in Table 2.
UPO reactions and AAO/UPO cascade
For the desired reaction to proceed further (from MMFA to FDCA), it was necessary to find a catalyst that could cleave the ether bond of the methoxyl group, to hydroxylate the 5-formylfurancarboxylic acid (FFCA) molecule and, if the ether cleavage left a hydroxyl group in the molecule, to oxidize the 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) molecule to FFCA (steps 2/5 or 3–5, respectively, in Fig. 1 scheme). In this regard, the UPO from Agrocybe aegerita has been reported to cleave a variety of ether bonds [20], as well as to hydroxylate FFCA to FDCA in the presence of H2O2 [13].
To clarify the enzymatic pathway, UPO (5 µM) was incubated with MMFA (1.5 mM) in the presence of H2O2 (1.5 mM, final concentration). Detection of FFCA as the sole product (data not shown) revealed that UPO is indeed capable to cleave the ether bond of MMFA while forming an additional carbonyl group in the molecule. This suggests that the reaction mainly proceeds through step 2 (Fig. 1 scheme), rather than through steps 3 and 4, although traces of HMFCA were detected as well. Therefore, only steps 1, 2, and 5 in the Fig. 1 scheme would be required for the production of FDCA from MMF, which is advantageous, since it saves one catalytic step that would require additional H2O2.
Given the two enzymatic activities described above—AAO’s ability to catalyze step 1 producing one equivalent of H2O2 and UPO being capable of catalyzing steps 2 and 5 consuming H2O2—an enzymatic cascade was assembled. AAO and UPO, both at a final concentration of 5 µM, reacted with 1.5 mM MMF (substrate/enzyme ratio of 300). This MMF concentration was selected to limit the UPO inhibition by H2O2 excess discussed below, although substrate saturation of AAO was not attained. Analysis of the reaction products revealed that, after 40 h, the reactions concluded—probably due to H2O2 depletion—resulting in approximately 25% conversion of initial MMF into FDCA (Fig. 3) together with 75% of MMFA (data not shown), which proved to be the main intermediate of the process and the bottleneck of the whole cascade.

Comparison of FDCA production (as mole percentage of initial MMF) in: (i) AAO/UPO cascade (blue line); (ii) AAO/UPO cascade supplemented with added H2O2 (orange line); (iii) AAO/UPO/MOX cascade (red line); and (iv) AAO/UPO/MOX cascade supplemented which methanol (after 72 h of reaction). See Fig. 2 for enzyme activities and substrate concentrations
Addition of exogenous H2O2 to the cascade
To test if the limited FDCA yields of the AAO/UPO cascade were due to the depletion of H2O2, this stoichiometric UPO substrate was added at different times after 24 h of reaction (up to 1.5 mM) and the products were analyzed during the subsequent incubation. Analysis of the reaction products showed that the whole reaction was improved upon the addition of H2O2. The FDCA yield corresponded to 40% conversion of initial MMF and the remaining 60% accounted for MMFA, while only traces of FFCA were detected (Figs. 2b and 3). Although the concentration of H2O2 (exogenously added and in situ produced by AAO) may have been sufficient, the complete conversion of MMF into FDCA was not achieved.
UPO showed activity during the whole process, displaying robustness as a biocatalyst by maintaining high levels of residual activity throughout the reaction (Fig. 2b, dashed line). UPO half-life and other catalytic performance parameters (TON, TOF, and TTN) under the assayed conditions are shown in Table 2, together with those of AAO.
Improvement of FDCA yield by MOX (and methanol) addition
The peroxygenase activity of UPO enables it to insert one O atom, which leads to ether breakdown, concomitantly with the formation of a carbonyl group in one of the products and a hydroxyl group in the other product [20]. As described above, the product of the reaction of MMFA with UPO was FFCA, in which a new carbonyl group was introduced. Consequently, UPO would release methanol as second fission product of the ‘quasi-benzylic’ peroxygenation reaction.
Therefore, with the aim of producing additional H2O2 to fuel the UPO reactions, commercially available MOX from Pichia pastoris was added to the enzymatic cascade described above to a final concentration of 1 µM (substrate/enzyme ratio of 1500). In this case, MOX catalyzes its canonical reaction, the oxidation of methanol to methanal and concomitantly, the reduction of O2 to H2O2. GC–MS analysis showed that upon the addition of MOX and the resulting increase of available H2O2, the UPO conversion was enhanced and FDCA yield reached 70% of the initial MMF concentration (Figs. 2c and 3). According to the analysis of products, the rate-limiting step of the UPO reactions (step 2 in Fig. 1 scheme) was demethoxylation, since MMFA was always the most abundant intermediate (up to 80% of initial MMF, after 24 h) compared to smaller amounts of FFCA (around 10% of the applied MMF).
To determine whether the improved FDCA yield (70%) was still limited by the amount of H2O2 available, exogenous methanol (1 mM final concentration) was added to the reaction after 72 and 96 h. In fact, with the addition of methanol (and subsequent H2O2 production), the FDCA formation further increased (Figs. 2d and 3), suggesting that the reaction was not limited by the activity of the biocatalyst. Thus, the FDCA conversion rose to 98% after 120 h, indicating that the limiting factor of the whole enzymatic cascade was the H2O2 availability. A summary of the conversion yields of MMF into MMFA, FFCA, and FDCA during operation (0–120 h) of the methanol-supplemented AAO/UPO/MOX cascade is provided in Table 3.
Discussion
AAO/UPO cascade for MMF oxidation
There is a wealth of oxidases involved in lignocellulose decay [21] that are potential biocatalysts for industry and are worth being studied [22]. In this work, the activity of P. eryngii AAO on lignocellulose-derived MMF is reported for the first time, which further widens/improves the application potential of AAO in FDCA production. In addition to fungal AAOs [13, 23, 24], other oxidases have proved to be suitable catalysts for the oxidation of HMF to FFCA or even FDCA employing O2 as co-substrate, as the so-called HMF oxidase (an intracellular bacterial enzyme from the same superfamily as AAO) [14, 15, 25].
MMF, which comes from the same renewable resources as HMF, appears as a better substrate for the synthesis of renewable polyesters, since it produces less dehydration by-products and displays higher stability upon storage. In the enzymatic oxidation of MMF, the drawback of AAO is its inability to produce the desired final product, FDCA. Acting alone, it lacks the ability to: (i) cleave the methoxy group in the MMF molecule and (ii) oxidize the carbonyl group in FFCA to FDCA. Nevertheless, the application of AAO in such process has the advantage that O2 is the only necessary reactant (co-substrate), apart from the chemical to be oxidized (substrate), to trigger the reaction, as it has been shown here. The production of H2O2 by AAO can be exploited for the creation of enzymatic cascades, in which another enzyme (peroxidase or peroxygenase) that uses it as electron acceptor (co-substrate) can be applied as downstream catalysts [13, 26]. Moreover, this is also a ‘smart’ way of destroying H2O2, which is an undesired by-product in terms of enzymatic performance and stability, forming H2O.
In the above context, the ability of UPO to perform an overwhelming number of different reactions [17, 27] allowed its application in the enzymatic synthesis of FDCA, not only from HMF [13] but also from MMF as shown in the present work. Here, it is shown that UPO is able to catalyze the cleavage of the ether bond in the MMF molecule thanks to the H2O2 produced by AAO. Furthermore, the fact that it catalyzed mainly the formation of a new carbonyl group in the molecule during ether fission (giving rise to FFCA from MMFA) saves one step in the enzymatic cascade designed, thereby reducing the need for H2O2 input. In general, the oxidation of MMF by AAO and UPO seems to proceed more efficiently than that of HMF [13, 28], due to the different polarity/reactivity of the methyl-ether functionality and the primary alcohol group, respectively.
Although the catalytic performances of the two biocatalysts are modest, with TTN values lower than 104 and kcat values lower than 1 s−1, the AAO/UPO cascade represents a good starting point for further improvement. The optimization of substrate and enzyme concentrations, required for an industrial exploitation of the cascade, would result in higher catalytic performance parameters.
The fact that UPO must catalyze two reactions (steps 2 and 5 in Fig. 1 scheme), whereas AAO does only one (step 1), causes a shortage in H2O2, so that the desired reactions cannot be completed (Fig. 3, blue dotted line). To solve this limitation, a three-member enzymatic cascade was developed as discussed below.
By-product oxidation to fuel the reaction
The addition of a third biocatalyst, MOX demonstrated that the limited FDCA yield (< 40% in the two-member AAO/UPO cascade) can be overcome by the in situ production of additional H2O2 to be used by UPO. Thus, a conversion of 70% from MMF to FDCA was attained using a three-enzyme cascade (AAO/UPO/MOX), which is fueled by the reduction of H2O2 (by UPO) and the oxidation of methanol (by MOX), two by-products generated by AAO and UPO, respectively. The respective results indicate that the 70% conversion is attained solely by the agents involved in the reaction as long as there is an atmospheric O2 input, as depicted in the three-member cascade scheme of Fig. 4.

Scheme of the three-enzyme (AAO/UPO/MOX) self-sufficient enzymatic cascade developed for the production of FDCA from MMF
The in situ self-generated fuel (H2O2 from the by-product methanol) here presented is a breakthrough in oxidative bioconversions. Other enzymatic cascades using UPO for oxygenations, in which methanol was applied as a sacrificial electron donor, have been reported, but the provenance of methanol was exogenous [26]. The production of methanal may be deleterious for enzymes if it reaches high concentrations, but such effect was not observed in the present study. The in situ generation of H2O2 has already been successfully applied in the bleaching industry using different flavoenzymes, such as glucose oxidase, alcohol oxidase [29] and cellobiose dehydrogenase/oxidase [30, 31].
Apart from improving the reaction yields, the cascade approach allows UPO to control the release of H2O2, since it is the enzyme itself that produces the methanol as substrate for MOX. It is well known that enzymes that bear heme groups, such as peroxidases and peroxygenases, are sensitive to high levels of their peroxide co-substrates resulting in deactivation via heme-bleaching. In fact, comparing the FDCA yields of the three-catalyst cascade with the cascade involving AAO and UPO to which H2O2 was added clearly indicates that UPO reactions proceed better if H2O2 is gradually supplied in situ by enzyme action (see red vs orange lines in Fig. 3). The addition of exogenous H2O2 provokes a sharp rise of its concentration that may lead to UPO inactivation. This results in a decrease of the enzyme’s half-life, thus lowering TTN, which negatively affects the degree of conversion into FDCA. Although complete MMF conversion was not achieved using the AAO/UPO/MOX cascade, the addition of a small amount of ‘extra methanol’ to the reaction mixture resulted in almost complete conversion (98%) of MMF into FDCA (Fig. 3). In fact, an excess of H2O2 (above the stoichiometric quantity required for the peroxygenation reaction) was necessary to overcome the reported catalase side-activity of UPO [32], which may consume some of the H2O2 by producing H2O and O2.
Conclusions
In this work, a completely enzymatic approach to produce FDCA from MMF is reported for the first time. The synergistic activities of AAO and UPO catalyze such conversion through an enzymatic cascade involving the two biocatalysts and O2 to trigger the reaction. UPO benefits from the H2O2 released by AAO to yield modest amounts of FDCA with H2O and methanol as by-products. The addition of MOX, oxidizing methanol under release of additional H2O2, critically improved the FDCA yield that was before limited by the amount of H2O2 produced by AAO. Thus, 70% conversion was achieved with the only involvement of the three biocatalysts, O2 and the by-products of the reaction, as illustrated in the Fig. 4 scheme. This yield could be further enhanced to 98% by the addition of some exogenous methanol resulting in additional H2O2 for UPO.
Methods
Reagents
P. pastoris MOX (EC 1.1.3.13), t-butyl-methyl-ether, MMFA, FDCA, N,O-bis(trimethylsilyl)-trifluoroacetamide (BSTFA) and 2H2O were purchased from Sigma-Aldrich (Saint Louis, MO, USA). MMF (= 5-[methoxymethyl]-2-furancarboxaldehyde) was bought from AK Scientific, Inc (Union City, CA, USA). FFCA was purchased from TCI America (Portland, OR, USA). AmplexRed® and HRP were obtained from Invitrogen (Walthem, MA, USA). H2O2 and DMSO-d6 were from Merck (Darmstadt, Hessen, Germany).
Enzyme production
AAO (EC 1.1.3.7) from the fungus P. eryngii was heterologously obtained from recombinant Escherichia coli W3110 harboring the pFLAG1 vector with the mature AAO cDNA (GenBank accession number AF064069). The enzyme was produced as inclusion bodies and further in vitro activated and purified as previously described [33].
PaDa-I variant of A. aegerita UPO (EC 1.11.2.1) was produced in P. pastoris, harboring the pPICZ-B-PaDa-I vector, grown in a 2-L glass fermentor. Expression was induced by the addition of methanol and the enzyme was chromatographically purified using Sepharose FF and Q-source columns (GE Healthcare, Piscataway, NJ, USA) as reported elsewhere [34, 35].
Kinetic studies
Kinetics of MMF, HMF, and DFF oxidation by AAO were studied by coupling the reaction of HRP and AmplexRed® at 25 °C, in 100 mM sodium phosphate, pH 7.0. H2O2 released by AAO is used by HRP to oxidize AmplexRed® to resorufin (Δε563 = 52,000 M−1 cm−1) in a 1:1 stoichiometric fashion. Therefore, spectrophotometric monitoring of the formation of colored resorufin allowed the indirect measurement of the AAO kinetic constants. Increasing concentrations of MMF (8–250 mM) were mixed with AAO (0.5 µM), AmplexRed® (0.06 mM), and HRP (24 µg mL−1) at a final volume of 1 mL. Reactions were triggered by addition of AAO and followed in a Cary 4000 spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Kinetics were obtained from the linear phase of resorufin production as change in absorbance over time and averaged data for each substrate concentration were fitted to Michaelis–Menten equation to obtain the kinetic parameters using SigmaPlot software (Systat Software Inc., San Jose, CA, USA).
Residual activities of AAO and UPO were measured after different times of incubation in the presence of MMF and its oxidized derivatives. AAO residual activity was determined by following spectrophotometrically the production of p-anisaldehyde (Δε285 = 16,950 M−1 cm−1) [36] from 200 µM p-methoxybenzyl alcohol, in 1 mL of 50 mM sodium phosphate, pH 6.0, at 25 °C. Regarding UPO, its residual activity was measured as the veratraldehyde (Δε310 = 9300 M−1 cm−1) [32] produced from 10 mM veratryl alcohol and 2 mM H2O2, in 1 mL of 100 mM sodium phosphate, pH 7.0, at 25 °C. Experimentally determined values of residual activity were fitted to Eq. 1 describing the enzymatic activity loss as a function of time. This allowed estimation of the half-lives (Eq. 2) of AAO and UPO, as well as their TTN (Eq. 3, considering the enzyme half-life), TON (Eq. 4) and TOF (Eq. 5):
MMF oxidation reactions
MMF reactions were performed in 100 mM sodium phosphate (pH 7.0) under continuous shaking at 200 rpm in a thermostated chamber at 28 °C. In all of them, the substrate was added to a final concentration of 1.5 mM, while UPO and AAO final concentration was 5 µM. MOX attained a final concentration of 1 µM. All enzymes and substrates were added from the beginning of the reaction in the different reaction mixtures employed, except H2O2 (1.5 mM final concentration) and methanol (1 mM final concentration), which were gradually added after different incubation times (24, 48 and 72 h, and 72 and 96 h, respectively).
GC–MS analyses
250-μL samples were harvested from the one-pot reactions after different times to analyze the products present. Reactions were stopped by adding HCl to give pH 2–3. Low pH values cause protonation of the organic acids and permit their liquid–liquid extraction, which was carried out by mixing the reaction mixtures with an excess of t-butyl-methyl-ether three times, followed by treatment with anhydrous NaSO4 to remove water traces. t-Butyl-methyl-ether was removed using a rotary evaporator at room temperature and samples were derivatized with 50 µL of BSTFA for 15 min at 25 °C [37].
Products were separated and identified using a gas chromatograph equipped with an HP-5MS column (Agilent, Santa Clara, CA, USA; 30 m × 0.25 mm internal diameter; 0.25 µm film thickness) coupled to a quadrupole mass detector. The oven program started at 110 °C (maintained for 2 min), increasing at 20 °C·min−1 until reaching 310 °C. Helium was used as the carrier gas at a flow rate of 1.2 mL min−1. The compounds involved in the MMF oxidative pathway were identified by comparing their mass spectra (and retention times) with those of derivatized authentic standards (Fig. 5). The following response factors were calculated as the slope of the fits of the responses of various concentrations of each standard compound (after its liquid–liquid extraction, derivatization and GC–MS analysis) to a linear equation: MMF: 1.7 × 107 total-ion mM−1; MMFA: 4.1 × 107 total-ion mM−1; FFCA: 3.1 × 107 total-ion mM−1; and FDCA: 3.6 × 107 total-ion mM−1. These response factors were used to estimate the mole percentage of each of the compounds in the reactions.

Mass spectra of authentic standards of the compounds involved in the cascade, as trimethylsilyl (TMS) derivatives. a MMF. b TMS-MMFA. c TMS-FFCA. d TMS-FDCA
NMR studies
1H-NMR was used to investigate the degree of hydration of the carbonyl group in the MMF molecule, using a Bruker 500 MHz instrument (Billenica, MA, USA). MMF (10 mM) was dissolved in 50 mM sodium phosphate (pD 7.0) prepared with 2H2O (99.9% isotopic purity). The internal reference for chemical shifts was the signal of the residual water proton (δH 4.9 ppm). Spectra of 10 mM MMF in DMSO-d6 (isotopic purity 99.98%) was run as a reference.
Abbreviations
- AAO:
-
aryl-alcohol oxidase
- BSTFA:
-
N,O-bis(trimethylsilyl)trifluoroacetamide
- DMSO-d6:
-
deuterated dimethylsulfoxide
- FDCA:
-
2,5-furandicarboxylic acid
- FFCA:
-
5-formylfurancarboxylic acid
- GC–MS:
-
gas chromatography–mass spectrometry
- HMF:
-
5-hydroxymethylfurfural
- HMFCA:
-
5-hydroxymethylfurancarboxylic acid
- HRP:
-
horseradish peroxidase
- k cat :
-
catalytic constant
- k cat/K m :
-
catalytic efficiency
- K m :
-
Michaelis constant
- MMF:
-
5-methoxymethylfurfural
- MMFA:
-
5-methoxymethylfurancarboxylic acid
- MOX:
-
methanol oxidase
- PEF:
-
poly(ethylene-furandicarboxylate)
- PET:
-
poly(ethylene-terephthalate)
- TOF:
-
turnover frequency
- TON:
-
turnover number
- TTN:
-
total turnover number
- UPO:
-
unspecific peroxygenase
References
Bozell JJ, Petersen GR. Technology development for the production of biobased products from biorefinery carbohydrates-the US Department of Energy’s “Top 10” revisited. Green Chem. 2010;12:539–54.
Papageorgiou GZ, Tsanaktsis V, Bikiaris DN. Synthesis of poly(ethylene furandicarboxylate) polyester using monomers derived from renewable resources: thermal behavior comparison with PET and PEN. Phys Chem Chem Phys. 2014;16:7946–58.
Gopalakrishnan P, Narayan-Sarathy S, Ghosh T, Mahajan K, Belgacem MN. Synthesis and characterization of bio-based furanic polyesters. J Polym Res. 2013;21:340.
de Jong E, Dam MA, Sipos L, Gruter G-JM. Furandicarboxylic acid (FDCA), A versatile building block for a very interesting class of polyesters. In: Smith P, editor. Biobased monomers, polymers and materials. Washington DC: ACS; 2012. p. 1–13.
Pellis A, Haernvall K, Pichler CM, Ghazaryan G, Breinbauer R, Guebitz GM. Enzymatic hydrolysis of poly(ethylene furanoate). J Biotechnol. 2016;235:47–53.
Balakrishnan M, Sacia ER, Bell AT. Etherification and reductive etherification of 5-(hydroxymethyl)furfural: 5-(alkoxymethyl)furfurals and 2,5-bis(alkoxymethyl)furans as potential bio-diesel candidates. Green Chem. 2012;14:1626–34.
Chen PX, Tang Y, Zhang B, Liu R, Marcone MF, Li X, Tsao R. 5-Hydroxymethyl-2-furfural and derivatives formed during acid hydrolysis of conjugated and bound phenolics in plant foods and the effects on phenolic content and antioxidant capacity. J Agric Food Chem. 2014;62:4754–61.
Douša M, Gibala P, Brichác J, Havlícek J. The formation of furfural compounds in selected saccharide- and polysaccharide-based pharmaceutical excipients. J Pharm Sci. 2012;101:1811–20.
Pacheco JJ, Davis ME. Synthesis of terephthalic acid via Diels-Alder reactions with ethylene and oxidized variants of 5-hydroxymethylfurfural. Proc Natl Acad Sci. 2014;111:8363–7.
Muñoz de Diego C, Schammel WP, Dam MAGGJM. Method for the preparation of 2,5-furandicarboxylic acid and esters thereof. PCT/NL2010/050653(WO/2011/043660). 14-4-2011. The Netherlands. 6-10-2010.
Janka ME, Lange DM, Morrow MC, Parker KR, Shaikh A, Partin LRJJC, Moody P, Shanks TE, Sumner CEJr. An oxidation process to produce a crude and/or purified carboxylic acid product. PCT/US2012/037204(WO/2012/161967). 29-11-2012. United States. 10-5-2012.
Mazoyer E, De Sousa Dias ASV, McKay B, Baars HJ, Vreeken VPC, Gruter GJM, Sikkenga DL. Process for the preparation of 2,5-furandicarboxylic acid. PCT/NL2014/050211(WO/2014/163500). 9-10-2014. The Netherlands. 4-4-2014.
Carro J, Ferreira P, Rodríguez L, Prieto A, Serrano A, Balcells B, Ardá A, Jiménez-Barbero J, Gutiérrez A, Ullrich R, Hofrichter M, Martínez AT. 5-Hydroxymethylfurfural conversion by fungal aryl-alcohol oxidase and unspecific peroxygenase. FEBS J. 2015;282:3218–29.
Dijkman WP, Groothuis DE, Fraaije MW. Enzyme-catalyzed oxidation of 5-hydroxymethylfurfural to furan-2,5-dicarboxylic acid. Angew Chem. 2014;126:6633–6.
Dijkman WP. HMF oxidase. Characterization, application and engineering of 5-(hydroxymethyl)furfural oxidase. Groningen: PhD thesis, Rijksuniversiteit; 2015.
Hernández-Ortega A, Ferreira P, Martínez AT. Fungal aryl-alcohol oxidase: a peroxide-producing flavoenzyme involved in lignin degradation. Appl Microbiol Biotechnol. 2012;93:1395–410.
Hofrichter M, Kellner H, Pecyna MJ, Ullrich R. Fungal unspecific peroxygenases: heme-thiolate proteins that combine peroxidase and cytochrome P450 properties. Adv Exp Med Biol. 2015;851:341–68.
Ozimek P, Veenhuis M, van der Klei IJ. Alcohol oxidase: a complex peroxisomal, oligomeric flavoprotein. FEMS Yeast Res. 2005;5:975–83.
Ferreira P, Hernández-Ortega A, Herguedas B, Rencoret J, Gutiérrez A, Martínez MJ, Jiménez-Barbero J, Medina M, Martínez AT. Kinetic and chemical characterization of aldehyde oxidation by fungal aryl-alcohol oxidase. Biochem J. 2010;425:585–93.
Kinne M, Poraj-Kobielska M, Ralph SA, Ullrich R, Hofrichter M, Hammel KE. Oxidative cleavage of diverse ethers by an extracellular fungal peroxygenase. J Biol Chem. 2009;284:29343–9.
Ferreira P, Carro J, Serrano A, Martínez AT. A survey of genes encoding H2O2-producing GMC oxidoreductases in 10 polyporales genomes. Mycologia. 2015;107:1105–19.
Martínez AT, Ruiz-Dueñas FJ, Camarero S, Serrano A, Linde D, Lund H, Vind J, Tovborg M, Herold-Majumdar OM, Hofrichter M, Liers C, Ullrich R, Scheibner K, Sannia G, Piscitelli A, Pezzella C, Sener ME, Kýlýç S, van Berkel WJH, Guallar V, Lucas MF, Zuhse R, Ludwig R, Hollmann F, Fernández-Fueyo E, Record E, Faulds CB, Tortajada M, Winckelmann I, Rasmussen J-A, et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol Adv. 2017;35:815–31.
Feldman D, Kowbel DJ, Glass NL, Yarden O, Hadar Y. Detoxification of 5-hydroxymethylfurfural by the Pleurotus ostreatus lignolytic enzymes aryl alcohol oxidase and dehydrogenase. Biotechnol Biofuels. 2015;8:63.
Hanke PD. Enzymatic oxidation of hydroxymethylfurfural. Patent (USA) 2012, 8,183,020 B2.
Dijkman WP, Fraaije MW. Discovery and characterization of a 5-hydroxymethylfurfural oxidase from Methylovorus sp. strain MP688. Appl Environ Microbiol. 2014;80:1082–90.
Ni Y, Fernández-Fueyo E, Baraibar AG, Ullrich R, Hofrichter M, Yanase H, Alcalde M, van Berkel WJH, Hollmann F. Peroxygenase-catalyzed oxyfunctionalization reactions promoted by the complete oxidation of methanol. Angew Chem Int Ed. 2016;55:798–801.
Wang Y, Lan D, Durrani R, Hollmann F. Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges? Curr Opin Chem Biol. 2017;37:1–9.
Karich A, Ullrich R, Hofrichter M. Enzymatic preparation of 2,5-furandicarboxylic acid (FDCA)—a substitute of terephthalic acid—by the joined action of three fungal enzymes. Microorganisms. 2018;6:5.
Lopez C, Cavaco-Paulo A. In-situ enzymatic generation of hydrogen peroxide for bleaching purposes. Eng Life Sci. 2008;8:315–23.
Pricelius S, Ludwig R, Lant NJ, Haltrich D, Guebitz GM. In situ generation of hydrogen peroxide by carbohydrate oxidase and cellobiose dehydrogenase for bleaching purposes. Biotechnol J. 2011;6:224–30.
Pricelius S, Ludwig R, Lant N, Haltrich D, Guebitz GM. Substrate specificity of Myriococcum thermophilum cellobiose dehydrogenase on mono-, oligo-, and polysaccharides related to in situ production of H2O2. Appl Microbiol Biotechnol. 2009;85:75–83.
Ullrich R, Nuske J, Scheibner K, Spantzel J, Hofrichter M. Novel haloperoxidase from the agaric basidiomycete Agrocybe aegerita oxidizes aryl alcohols and aldehydes. Appl Environ Microbiol. 2004;70:4575–81.
Ruiz-Dueñas FJ, Ferreira P, Martínez MJ, Martínez AT. In vitro activation, purification, and characterization of Escherichia coli expressed aryl-alcohol oxidase, a unique H2O2-producing enzyme. Protein Express Purif. 2006;45:191–9.
Molina-Espeja P, Garcia-Ruiz E, Gonzalez-Perez D, Ullrich R, Hofrichter M, Alcalde M. Directed evolution of unspecific peroxygenase from Agrocybe aegerita. Appl Environ Microbiol. 2014;80:3496–507.
Molina-Espeja P, Ma S, Maté DM, Ludwig R, Alcalde M. Tandem-yeast expression system for engineering and producing unspecific peroxygenase. Enzyme Microb Technol. 2015;73–74:29–33.
Ferreira P, Medina M, Guillén F, Martínez MJ, van Berkel WJH, Martínez AT. Spectral and catalytic properties of aryl-alcohol oxidase, a fungal flavoenzyme acting on polyunsaturated alcohols. Biochem J. 2005;389:731–8.
Teixidó E, Santos FJ, Puignou L, Galceran MT. Analysis of 5-hydroxymethylfurfural in foods by gas chromatography-mass spectrometry. J Chromatogr A. 2006;1135:85–90.
Authors’ contributions
JCarro performed the biocatalytic experiments and wrote the manuscript. EFF produced and purified the UPO used in the experiments. CFA and JCañada performed the NMR experiments. RU and MH discovered UPO. MA engineered the UPO variant for yeast expression. PF and ATM designed the experiments and revised the results and manuscript. All the authors revised the final version of the manuscript, and significantly contributed to the discussion and conclusions obtained. All authors read and approved the final manuscript.
Acknowledgements
JCarro acknowledges an FPU (Formación del Profesorado Universitario) Fellowship (AP2012-2041) awarded by the Spanish Ministry of Education, Culture and Sports. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Competing interests
Patent with reference number P201730805 was filed on 16th June 2017 that involves part of the work herein described.
Consent for publication
Not applicable
Ethical approval and consent to participate
Not applicable
Funding
This work has been funded by the H2020 BBI-JU (http://www.bbi-europe.eu) project EnzOx2 (H2020-BBI-PPP-2015-2-720297) together with the INDOX EU project (FP7-KBBE-2013-7-613549) and the NOESIS (BIO2014-56388-R) and GENOBIOREF (BIO2017-86559-R) projects of the Spanish Ministry of Economy, Industry and Competitiveness, co-financed by FEDER funds.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carro, J., Fernández-Fueyo, E., Fernández-Alonso, C. et al. Self-sustained enzymatic cascade for the production of 2,5-furandicarboxylic acid from 5-methoxymethylfurfural. Biotechnol Biofuels 11, 86 (2018). https://doi.org/10.1186/s13068-018-1091-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-018-1091-2