
Abstract
This article is one of ten reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2018. Other selected articles can be found online at https://www.biomedcentral.com/collections/annualupdate2018. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901.
Similar content being viewed by others


Introduction
In recent years, the surge of culture-independent methods to study bacterial populations has led to an increasing body of evidence pointing towards the microbiome as an important player in the pathophysiology of a whole spectrum of diseases that affect the critically ill, including trauma and sepsis [1,2,3,4]. Techniques such as 16S rRNA and shotgun metagenomic sequencing have opened up a new area of research, enabling detailed investigations of complex populations of bacteria and their effects on health and disease [5, 6]. Some have even called the microbiome a separate organ given its numerous roles in metabolism, development of the immune system and host defense against pathogens [7].
Microbiota is an overarching term for all the microbes in a population, consisting of bacteria, archaea and eukarya [8]. Most studies that have been performed look at the bacterial microbiota because of its high abundance and high diversity. The collective of microbes in a population is referred to as the microbiota and the genetic content as the microbiome.
The real value of all this novel knowledge for the clinical care of patients on the intensive care unit (ICU) still has to be established. Nonetheless, data are accumulating that underscore the potential importance of the microbiome for intensive care medicine. On any given day, three-fourths of all patients on the ICU are treated with antibiotics, which are known to cause severe collateral damage to the microbiome [9]. Besides antibiotics, there are multiple external modulators of the gut microbiota applied during the clinical care of patients on the ICU, such as gastric acid inhibition, the route of feeding, sedatives and opioids [3, 10]. Novel strategies are being designed to intervene on the microbiome to prevent or treat trauma and sepsis. Excitingly, a very recent randomized, placebo-controlled trial among 4556 healthy infants in India showed that the oral administration of Lactobacillus plantarum in combination with fructooligosaccharide in the first week of life could reduce the occurrence of sepsis in the first 60 days of life [4]. Other evidence points towards the use of synbiotics as an adjunctive therapy to prevent postoperative complications, such as surgical site infections and sepsis among adult surgical patients [11]. Most research in this field has focused on the intestinal microbiome; however, current research is also starting to show the importance of the lung microbiome for ICU patients [1]. For example, enrichment of the lung microbiome with gut bacteria seems to play a role in the pathogenesis of acute respiratory distress syndrome (ARDS) [12].
In this chapter, we will discuss the emerging role of the bacterial microbiotas in the gut and the lung in the critically ill. First, we will discuss the techniques available to study the bacterial microbiome, then we will continue into the gut and lung microbiome and end with some key questions for future research in the field of microbiota-targeted therapies on the ICU.
Microbiota analyses
The composition of the microbiota in the gut has been studied extensively using 16S ribosomal RNA (rRNA) gene-targeted approaches. The use of this single genetic marker has revolutionized microbial ecology [13, 14]. It has become relatively easy to amplify the 16S rRNA encoding genes from environmental DNA. Nowadays, with next-generation sequencing techniques, many microbial environments can be studied in depth and at high resolution in space and time, using relatively straightforward procedures.
To address not only microbiota composition, but rather focus on the metabolic potential and actual activity of the intestinal microbiota, “meta’omic” approaches have emerged during the last decade and are now widely used [15,16,17]. Each of the meta’omic approaches provides different information about the functional potential or activity profiles of a microbial community.
Metagenomics is used to determine the collective genomes of members present in a microbial community as well as their functional capacity. Metagenomics was used in the Meta HIT (Metagenomics of the Human Intestinal Tract) project and provided a human microbiome-derived gene catalogue with over 3 million genes, indicating a community of over 150 species in an individual and a 100-fold larger non-redundant gene set compared to the human gene complement [16].
Metatranscriptomics and metaproteomics are able to provide information about the functions expressed by the members of the community. For example, metaproteomics analysis in healthy humans revealed a difference in the amount of proteins expressed and the proteins predicted by metagenomics. Moreover, metaproteomics in fecal material not only gives information on the bacterial proteins, but also on the major human proteins, giving insights into the main host responses [18]. However, it is difficult and sometimes not possible to use conserved proteins to distinguish between species or even higher taxonomic levels (e.g., genus or family). Consequently, by using this technique, you lose some of the taxonomic information otherwise gained by using metagenomics, metatranscriptomics or targeted 16S rRNA sequencing.
Metatranscriptome analysis of the gastrointestinal tract microbiota enables elucidation of the specific functional roles microbes have in this complex community. Although initial studies on the human large intestine revealed that different functions are expressed among individuals, core functions of the microbiota appear to be consistently expressed in different individuals [19, 20]. Moreover, metatranscriptome analyses of the small intestinal microbiota underpinned the cross-feeding between two dominant members of the small intestinal microbiota, i.e., Streptococcus spp. and Veilonella spp., in which the lactate produced by Streptococcus spp. is used as a carbon and energy source by the Veillonella spp. [17]. Metatranscriptome analysis of the microbiota in humanized mice revealed that mice colonized with the microbiota obtained from a lean human donor had greater expression of genes involved in polysaccharide breakdown and in propionate and butyrate production as compared to those colonized with the microbiota of an obese human donor [21]. These findings imply that metatranscriptomics can provide insight into the differential activity profiles in the intestinal microbiota, and enables reconstruction of the metabolic activity profile of microbial communities.
Metabolomic approaches are used to detect and quantify the metabolites that are produced by the microbial community. This approach has been suggested to be applicable as a diagnostic tool in diseases that involve aberrations of the intestinal microbiota composition and activity [22]. However, as with metaproteomics, you also lose information on the specific bacteria producing these metabolites. To overcome the loss of taxonomic information in metaproteomics and metabolomics, these techniques are often combined with 16S rRNA sequencing, metagenomics or metatranscriptomics.
These tools often generate very complex datasets with many different measurements (e.g., species or function) under various conditions. Therefore, these multivariate meta’omics datasets need tools to simplify the datasets and focus on correlations between points of interest, such as dietary interventions and the bacterial community or the bacterial community and host responses. Multivariate statistics are used to handle these large datasets and enable a relatively quick focus on data of importance [23]. An overview of available techniques to analyze the microbiome is provided in Fig. 1.

Overview of techniques to detect bacterial microbiota. To detect which bacterial species there are in a sample, there are three options: 1) use quantitative polymerase chain reaction (qPCR) to detect total bacterial 16S rRNA gene and/or in combination with a specific qPCR for a bacterial group or species; 2) use 16S rRNA gene for amplicon multiplex sequencing to get information on the taxonomic distribution in a sample; 3) use total DNA of a sample for shot-gun sequencing of the metagenomics content to get information on taxonomic distribution and functions, examples of which are indicated in the box, to show that you have functional information within a taxonomic group, in this case butyrate kinase and glycoside hydrolase. The dollar sign below the different techniques is an indicator of the price: depending on the sample type and information depth, prices are variable, but, roughly, a one-dollar sign is around 5–10 dollars per sample; the two-dollar sign is 50–150 dollars per sample, and the three-dollar sign is 300–1000 dollars per sample
The gut microbiota
Human individuals can harbor over 150 different microbial species in their gut, which collectively encode more than 100-fold more non-redundant genes than there are in the human genome [16, 24]. More recent data, however, has challenged this number, suggesting that the ratio between bacteria and human cells is closer to 1:1 [25]. In healthy humans, the intestinal microbiota consists of members of all three domains of life: bacteria, archaea and eukarya, of which the bacterial community is the most abundant and diverse [8]. Nine different bacterial phyla have been recorded in humans so far, of which the bacteroidetes and firmicutes dominate [8, 16, 24]. Many of these bacteria in the gut have not been cultivated. Recent breakthroughs in the successful culture of the previously ‘unculturable’ human microbiota have revealed a whole spectrum of novel bacterial species and taxa [26]. The application of novel sequencing techniques provides an opportunity to understand this complex ecosystem much better [8, 13].
The intestinal microbiota plays a critical role in priming the host’s immune system, gut maturation and gut functions, such as nutrient uptake and metabolism, mucosal barrier function, enteric nervous system and motility [27,28,29]. Numerous host genes seem to be specifically altered in response to various members of the microbiota, showing the importance of the microbial composition to the body’s response [30, 31].
Modulators of the microbiota on the ICU
The healthy intestinal microbiota is affected primarily by the host and the diet. In critically ill patients, these two factors both play an important role. However, in the critically ill it is also important to note that pathogens are able to outcompete other members of the intestinal microbiota more easily than in healthy humans, also changing the microbial composition. These pathogens benefit from a changed environment in critically ill patients, which are (though not per se all at the same time) decreased transit time, lack of ‘normal’ nutrition, oxygen levels and antibiotic usage [1]. Most of these opportunistic pathogens, except for Clostridium difficile infections, are Gram-negative aerobic species belonging to the proteobacteria, which thrive under the changed environment in critically ill patients. The universal use of antibiotics is probably the main cause of the severe bacterial dysbiosis seen in patients admitted to the ICU [32,33,34,35,36]. In addition, because of no or limited nutritional intake by patients on the ICU, nutrition for the microbiota in the intestine also decreases, affecting the microbiota composition. For example, in the large intestine, the microbiota ferments non-digested dietary fibers and dietary and host-derived proteins. The fermentation of fibers is necessary for the production of butyrate by the microbiota, which epithelial cells use as an energy source. The study of the clinical relevance of these effects in critically ill patients is, however, still in its infancy.
The potential clinical relevance of the gut microbiota on the ICU
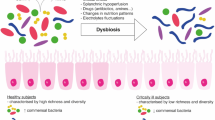
An imbalance in the homeostasis or ‘dysbiosis’ of the gut microbiota has been associated with a range of different diseases, including diabetes, obesity, inflammatory bowel disease and rheumatoid arthritis [5, 6]. This association with disease has led to investigations into the gut microbiota’s involvement at the systemic level. In the critically ill, the intestinal microbiota has been analyzed in a couple of studies so far. In general, we can state that critically ill patients admitted to the ICU have a state of dysbiosis of the intestinal microbiota [32,33,34]. Figure 2 shows a schematic representation of the composition of the intestinal microbiota in the critically ill in comparison to a healthy human. Overall, the intestinal microbiota of critically ill patients admitted to the ICU with sepsis is characterized by lower diversity, lower abundance of key commensal genera (such as Faecalibacterium, Blautia, Ruminococcus), and in some cases overgrowth (over 50% relative abundance) by one genera, such as Escherichia/Shigella, Salmonella, Enterococcus, C. difficile or Staphylococcus [32,33,34,35,36].

Intestinal microbiota in health and critical illness. The microbiota in a healthy individual is a diverse ecosystem with beneficial and commensal bacteria and low abundances of opportunistic pathogens, which are not harmful in small numbers. As a result of certain therapies, such as antibiotics, the microbial composition can be disrupted. The intestinal microbiota of critically ill patients is less diverse and contains more opportunistic pathogens and less beneficial and commensal bacteria
A normal, healthy intestinal microbiota protects against an invasion of pathogens like Enterococcus faecium, Escherichia coli and C. difficile. It comes as no surprise that severe infections caused by these pathogens often occur in patients with a recent history of antibiotic use. The microbiota of these patients is therefore probably disrupted, which allows for the overgrowth of antibiotic-resistant pathogenic and opportunistic bacteria that are frequently encountered in hospital settings [37].
In sepsis, the focus often lies on the identification of a single pathogen as the causative agent. However, there is an increasing realization that most pathogens do not act in isolation and that infections have ‘polymicrobial’ phenotypes and are thus linked to the status of the microbiota in the patient. The initial state of the microbiota can influence susceptibility to infection [38] and severity of infection [39]. Recent preclinical data derived from animal models suggest that the gut microbiome plays a protective role in the host defense against sepsis [39,40,41]. As an example, antibiotic induced disruption of the gut microbiome leads to increased inflammation and bacterial dissemination in murine models of both Gram-positive and Gram-negative pneumosepsis [39, 40]. ICU patient data have suggested that the loss of microbiome diversity could predict the length of stay of patients on the ICU further underscoring the potential clinical relevance of the gut microbiome for intensive care medicine [33].
The evolving role of the lung microbiota in critical illness
Healthy lungs were long thought to be sterile, until recent studies showed that bacteria can be cultured from healthy lungs [42]. The lung environment is less advantageous for bacteria to grow and flourish when compared to the intestine, leading to a less dense bacterial community compared to the gut. A lack of nutrients, the bidirectional movement of the lung, the aerobic environment, and the coating of alveoli with a lipid-rich surfactant that has bacteriostatic effects all contribute to this harsh environment for bacteria [42, 43]. Furthermore, obtaining a sample from the lung is far more invasive than obtaining a stool sample to study the microbiome. Contamination from the sampling device or from the upper respiratory tract is another point to take into consideration.
In a healthy lung, the microbiota is delicately balanced by the reproduction rate of present bacteria and the immigration and elimination rate of bacteria. Under normal circumstances, the reproduction rate of bacteria remains low and the immigration and elimination high. However, in critical illness, sedatives and endotracheal intubation can decrease the mucociliary clearance and cough reflex, leading to decreased microbial elimination. Furthermore, mechanical ventilation can cause an increase in alveolar edema, which can lead to an increase in available nutrients in the lung and areas where the oxygen levels are lower, allowing bacteria to thrive [1].
In healthy lungs, it is thought that most of the bacteria come from the oral microbiota. The lung microbiota most closely resembles the microbiota of the oropharynx, more so than that of the nasopharynx, gastrointestinal tract or inhaled air [42]. The healthy oropharynx contains benign Veillonella spp. and Prevotella spp., and, therefore, these are also found in healthy lungs. During critical illness, the oropharynx can become overpopulated with pathogenic proteobacteria, such as Pseudomonas aeruginosa and Klebsiella pneumoniae. Furthermore, in critical illness the stomach and small intestine can become the primary source of bacterial migration to the lung [1, 42].
ARDS and pneumonia are thought to cause alveolar injury and as a consequence induce changes in the microbiome. The alveoli become covered with protein-rich fluids and the bactericidal surfactant from the alveoli becomes inactivated, making the alveoli a more hospitable environment for bacteria [1]. Furthermore, Dickson et al. [44] proposed a theory regarding the existence of a positive feedback loop between the growth of certain bacterial species in the lung and the local inflammatory response; as the bacterial population grows, it starts to limit itself due to nutrient shortage while it also provokes an increased inflammatory response. This inflammatory response can consequently lead to endothelial and epithelial injury, releasing fluids that are rich in proteins and nutrients, thus stimulating bacterial growth. Further bacterial growth will increase the local inflammation, thus creating a positive feedback loop. This suggests that in some cases the body’s inflammatory response may be making the infection worse.
The link between the microbiota and the gut-lung Axis
The intestinal microbiota has emerged as a key component of both local and systemic immunity. Epithelial and immune cells gain information directly from bacteria and local cytokine responses and subsequently adjust inflammatory responses. Microbiome research combining the gut and lung has started to show an association between the composition of the intestinal microbiota and lung health [45, 46]. Experimental germ-free and antibiotic murine models have shown that the body’s microbiota is important in the defense against influenza and several types of bacterial pneumonia [46]. Moreover, probiotics containing Lactobacillus spp. and Bifidobacterium spp. have been shown to improve incidence and outcome of respiratory infections. Additionally, exposure to Toll-like receptor (TLR) agonists and nucleotide-binding oligomerization domain (NOD)-like receptors in the intestine, by substances such as lipopolysaccharide (LPS), peptidoglycan and lipoteichoic acid, has been shown to increase the lungs’ ability to clear bacteria [45]. These studies suggest that the intestinal microbiota is important for airway defenses. Moreover, human and murine studies have shown that the lungs can contain gut-associated bacteria during sepsis and ARDS. However, this gut-lung axis does not seem to be a one-way street. Pulmonary infection with tuberculosis, influenza and Burkholderia pseudomallei have all been shown to have a significant effect on the composition of the gut microbiome in murine models [40, 47, 48]. Figure 3 shows a schematic overview of the potential consequences of microbial dysbiosis in the critically ill in both the lung and intestine.

The gut and lung microbiota in critical illness. A healthy gut microbiome plays a protective role in host defense against local and pulmonary pathogen invasion. In the critically ill, there is often dysbiosis in the lung and intestinal microbiotas, which can contribute to diseases like pneumonia and acute respiratory distress syndrome (ARDS)
The road ahead
Hospital-acquired infections are a huge problem in patients on the ICU. Antibiotic administration is the usual form of treatment or prevention of these infections and is lifesaving. However, as a side-effect, it severely affects the microbiota, risks induction of antibiotic resistant pathogens and potentially even the new onset of severe infection. Restoring the microbiota after antibiotic treatment has the potential to reduce infections [10, 23]. Different approaches can be utilized: probiotics, prebiotics (a dietary ingredient that promotes beneficial microbiota) and synbiotics (a combination of a probiotic with a prebiotic) have all been tested in various patient groups on the ICU with mixed results [4, 49]. Probiotics will not reestablish a complete microbiota. This can be achieved to a larger extent by performing a fecal microbiota transfer (FMT). FMT has been used successfully to restore re-occurring C. difficile infections in patients on the ICU. The first case reports on the successful use of FMT in patients with therapy-refractory sepsis have been published [10]. A key factor, however, will be establishing the exact mechanism by which certain components of the gut and/or lung microbiome play their presumed protective effects on the health of patients admitted to the ICU. This could enable us to develop targeted therapies to restore the microbiome in this very vulnerable patient population.
At the present time, we lack information on the dynamics of the microbiota in the critically ill in order to develop targeted therapies to restore the microbiome. There is a need for information on how the microbiota restores with and without interference or therapies in the critically ill. Moreover, these patients should be followed for long-term periods to monitor re-occurring infections. This information will aid in developing targeted approaches, such as the use of probiotics or FMT, to restore the microbiota and prevent infections during a patient’s recovery.
Conclusion
The importance of the intestinal and lung microbiotas is often overlooked on the ICU. Currently, we can explore the microbiome using a vast array of techniques, giving us ‘meta’ libraries of data, which has allowed researchers to show the potential crucial role of the microbiota for ICU patients. During a patient’s stay on the ICU, their microbiota is influenced by both their illness and the care provided. For example, the gut microbiota of critically ill patients admitted to the ICU with sepsis is characterized by lower diversity, lower abundance of key commensal genera and, in some cases, an overgrowth by one genera. These changes in the microbiota can, in turn, affect patient outcome and susceptibility to infection. Furthermore, the lung microbiota has only recently been shown to be important in the critically ill. Intriguingly, recent evidence also points to a bi-directional microbiota-mediated role between the gut and the lung, the so called gut-lung axis. The importance of these microbiotas is pushing us towards new types of treatments, in which we also start to treat the microbiota.
Acknowledgements
This work was supported by the Netherlands Organization for Scientific Research (NWO), the Horizon2020 Marie Skłodowska-Curie International Training Network “the European Sepsis Academy” and The Netherlands Organization for Health Research development (ZonMw).
References
Dickson RP. The microbiome and critical illness. Lancet Respir Med. 2016;4:59–72.
Baumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535:85–93.
Haak BW, Wiersinga WJ. The role of the gut microbiota in sepsis. Lancet Gastroenterol Hepatol. 2017;2:135–43.
Panigrahi P, Parida S, Nanda NC, et al. A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature. 2017;548:407–12.
Blaut M, Clavel T. Metabolic diversity of the intestinal microbiota: implications for health and disease. J Nutr. 2007;137(3 Suppl 2):751S–5S.
Guarner F. Enteric flora in health and disease. Digestion. 2006;73(Suppl 1):5–12.
Baquero F, Nombela C. The microbiome as a human organ. Clin Microbiol Infect. 2012;18(Suppl 4):2–4.
Rajilic-Stojanovic M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047.
Vincent J-L, Rello J, Marshall J, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302:2323–9.
Haak BW, Levi M, Wiersinga WJ. Microbiota-targeted therapies on the intensive care unit. Curr Opin Crit Care. 2017;23:167–74.
Kasatpibal N, Whitney JD, Saokaew S, Kengkla K, Heitkemper MM, Apisarnthanarak A. Effectiveness of probiotic, prebiotic, and synbiotic therapies in reducing postoperative complications: a systematic review and network meta-analysis. Clin Infect Dis. 2017;64(suppl 2):S153–60.
Dickson RP, Singer BH, Newstead MW, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. 2016;1:16113.
Tringe SG, Hugenholtz P. A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol. 2008;11:442–6.
Pace NR, Sapp J, Goldenfeld N. Phylogeny and beyond: scientific, historical, and conceptual significance of the first tree of life. Proc Natl Acad Sci U S A. 2012;109:1011–8.
Fritz JV, Desai MS, Shah P, Schneider JG, Wilmes P. From meta-omics to causality: experimental models for human microbiome research. Microbiome. 2013;1:14.
Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Zoetendal EG, Raes J, van den Bogert B, et al. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012;6:1415–26.
Verberkmoes NC, Russell AL, Shah M, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3:179–89.
Franzosa EA, Morgan XC, Segata N, et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci U S A. 2014;111:E2329–38.
Gosalbes MJ, Durban A, Pignatelli M, et al. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One. 2011;6:e17447.
Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214.
De Preter V, Verbeke K. Metabolomics as a diagnostic tool in gastroenterology. World J Gastrointest Pharmacol Ther. 2013;4:97–107.
Martin FP, Wang Y, Sprenger N, et al. Top-down systems biology integration of conditional prebiotic modulated transgenomic interactions in a humanized microbiome mouse model. Mol Syst Biol. 2008;4:205.
Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–20.
Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164:337–40.
Browne HP, Forster SC, Anonye BO, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533:543–6.
Ciccia F, Guggino G, Rizzo A, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis. 2017;76:1123–32.
Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–7.
Backhed F, Roswall J, Peng Y, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690–703.
Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–4.
Leser TD, Molbak L. Better living through microbial action: the benefits of the mammalian gastrointestinal microbiota on the host. Environ Microbiol. 2009;11:2194–206.
Lankelma JM, van Vught LA, Belzer C, et al. Critically ill patients demonstrate large interpersonal variation in intestinal microbiota dysregulation: a pilot study. Intensive Care Med. 2017;43:59–68.
McDonald D, Ackermann G, Khailova L, et al. Extreme dysbiosis of the microbiome in critical illness. Msphere. 2016;1:e00199–16.
Wischmeyer PE, McDonald D, Knight R. Role of the microbiome, probiotics, and ‘dysbiosis therapy’in critical illness. Curr Opin Crit Care. 2016;22:347–53.
Zaborin A, Smith D, Garfield K, et al. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. MBio. 2014;5:e1361–14.
Ojima M, Motooka D, Shimizu K, et al. Metagenomic analysis reveals dynamic changes of whole gut microbiota in the acute phase of intensive care unit patients. Dig Dis Sci. 2016;61:1628–34.
Pamer EG. Resurrecting the intestinal microbiota to combat antibiotic-resistant pathogens. Science. 2016;352:535–8.
Yooseph S, Kirkness EF, Tran TM, et al. Stool microbiota composition is associated with the prospective risk of plasmodium falciparum infection. BMC Genomics. 2015;16:631.
Schuijt TJ, Lankelma JM, Scicluna BP, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut. 2016;65:575–83.
Lankelma JM, Birnie E, Weehuizen TAF, et al. The gut microbiota as a modulator of innate immunity during melioidosis. PLoS Negl Trop Dis. 2017;11:e5548.
Deshmukh HS, Liu Y, Menkiti OR, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia Coli K1 sepsis in neonatal mice. Nat Med. 2014;20:524–30.
Huffnagle GB, Dickson RP, Lukacs NW. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 2017;10:299–306.
Dickson RP, Huffnagle GB. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog. 2015;11:e1004923.
Dickson RP, Erb-Downward JR, Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med. 2014;2:238–46.
Budden KF, Gellatly SL, Wood DL. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15:55–63.
Marsland BJ, Trompette A, Gollwitzer ES. The gut-lung axis in respiratory disease. Ann Am Thorac Soc. 2015;12(Suppl 2):S150–6.
Winglee K, Eloe-Fadrosh E, Gupta S, Guo H, Fraser C, Bishai W. Aerosol mycobacterium tuberculosis infection causes rapid loss of diversity in gut microbiota. PLoS One. 2014;9:e97048.
Deriu E, Boxx GM, He X, et al. Influenza virus affects intestinal microbiota and secondary salmonella infection in the gut through type i interferons. PLoS Pathog. 2016;12:e1005572.
Manzanares W, Lemieux M, Langlois PL, Wischmeyer PE. Probiotic and synbiotic therapy in critical illness: a systematic review and meta-analysis. Crit Care. 2016;19:262.
Acknowledgements
Not Applicable.
Funding
Publication costs were funded by the Center for Experimental and Molecular Medicine (CEMM).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
Literature search (N.S.W.); writing (N.S.W., F.H., W.J.W.); overview of manuscript (W.J.W.). All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wolff, N.S., Hugenholtz, F. & Wiersinga, W.J. The emerging role of the microbiota in the ICU. Crit Care 22, 78 (2018). https://doi.org/10.1186/s13054-018-1999-8
Published:
DOI: https://doi.org/10.1186/s13054-018-1999-8