
Abstract
Background
Bardet–Biedl syndrome (BBS) is a rare inherited multisystemic disorder with autosomal recessive or complex digenic triallelic inheritance. There is currently no treatment for BBS, but some morbidities can be managed. Accurate molecular diagnosis is often crucial for the definition of appropriate patient management and for the development of a potential personalized therapy.
Methods
We developed a next-generation-sequencing (NGS) protocol for the screening of the 18 most frequently mutated genes to define the genotype and clarify the mutation spectrum of a cohort of 20 BBS Italian patients.
Results
We defined the causative variants in 60% of patients; four of those are novel. 33% of patients also harboured variants in additional gene/s, suggesting possible oligogenic inheritance. To explore the function of different genes, we looked for correlations between genotype and phenotype in our cohort. Hypogonadism was more frequently detected in patients with variants in BBSome proteins, while renal abnormalities in patients with variations in BBSome chaperonin genes.
Conclusions
NGS is a powerful tool that can help understanding BBS patients’ phenotype through the identification of mutations that could explain differences in phenotype severity and could provide insights for the development of targeted therapy. Furthermore, our results support the existence of additional BBS loci yet to be identified.
Similar content being viewed by others


Background
Bardet-Biedl syndrome (BBS) is a rare inherited, clinically and genetically heterogeneous, multisystemic ciliopathy with various primary and secondary clinical manifestations [1]. Although the common postaxial hexadactyly is evident at birth, in absence of a family history the diagnosis is usually made after the manifestation of ocular involvement. The main features are: retinal dystrophy (cone-rod type) often leading to blindness, upper and lower limbs polydactyly, early-onset truncal obesity, intellectual impairment, hypogonadism and renal abnormalities. A number of secondary features is also described [1]. Life expectancy can be reduced, mainly due to renal failure [2].
Twenty-one different loci (BBS1-BBS21) have been associated with this syndrome. The genes mostly code for proteins forming the core BBSome complex (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8 and BBS9) or part of a BBS chaperone complex (BBS6, BBS10 and BBS12) which plays an essential role in the stabilization and regulation of the BBSome [3, 4]. Other genes code for proteins with roles in the localization and activation of BBSome (ARL6) or BBSome entry into cilia (BBS17) or are associated with the BBSome complex (BBS14) (Fig. 1 and Additional file 1: Table S1). The functions of some of the proteins are not fully understood. BBSome is a stable protein complex that functions in the biogenesis and maintenance of the primary cilium (Fig. 1), a structure that is ubiquitously expressed and highly conserved through evolution, and in modulating ciliary protein trafficking. Primary cilia serve as sensors of the extracellular environment, they receive and transduce signals from light, chemical, or mechanical stimuli, [5] in addition they play a role in several signalling pathways important for development and tissue homeostasis, their alteration results in abnormalities and multiorgan disfunctions. Many different pathways have been linked to the primary cilium, including Hedgehog, Wnt, Notch, Hippo, GPCR, PDGF, mTOR, and TGF-beta [6].

Schematic representation of the cilium and intraflagellar transport. 1. transition zone; 2. basal body; 3. axoneme; 4. pericentriolar area. BBS-chaperonin complex (BBS6, BBS10, BBS12) binds and stabilizes the BBS protein to form the BBSome (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, BBS17, BBS18). BBSome plays a critical role in the regulation of cilia composition and in intraflagellar trafficking. Indeed, transmembrane (TM) and periferal membrane protein are transported in the cilium in a BBSome dependent manner. BBS3 triggers BBSome complex /cargo proteins interaction and their transition across the control barrier (transition zone - BBS13, BBS14, BBS15) into the cilium. In bold, genes included in our NGS panel
For many years BBS was considered an autosomal recessive disease, but recently evidence suggesting complex digenic triallelic inheritance has been described [7,8,9]. This could partly explain the large, phenotypic heterogeneity found in BBS patients, both inter- and intrafamilial [10].
There is currently no treatment for BBS, but some of the co-morbidities can be managed. Precise identification of the causative gene(s) is therefore a fundamental step toward a personalized therapeutic approach and management of genotype-related conditions [8, 11,12,13,14,15,16,17]. Thus, genetic analysis and accurate phenotyping are fundamental for stratifying patients and addressing appropriate therapy. In our study we analysed the mutation spectrum in a cohort of 20 Italian patients with BBS, investigating the tri-allelic hypothesis and analysing genotype-phenotype correlation.
Materials and methods
Patients
Twenty caucasian patients diagnosed in different hospital across Italy with Bardet-Biedl syndrome were retrospectively included in the study. The mean (±SD) age was 29 ± 17.1 (range 9–63) years and the male/female ratio was 13:7. The mean age at diagnosis was 5.4 ± 7.2 (range 0–24) years. No consanguinity in their families was reported [except for two probands that stated a distant kinship]. Clinical diagnosis of BBS was made according to the accepted criteria [1]. Genetic testing was performed on germline DNA extracted from either saliva or blood of the proband.
Mutation analysis
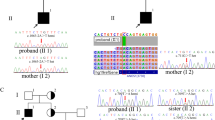
A custom-made oligonucleotide probe library was designed to capture all coding exons and flanking exon/intron boundaries (±15 bp) of 18 genes known to be associated with Bardet-Biedl syndrome (Additional file 1: Table S1 and Additional file 2: for protocol details). DNA from the proband was analysed. Identified variants with likely clinical significance (pathogenic, likely pathogenic and of unknown significance according to the ACMG guideline) [18] were confirmed by bidirectional Sanger sequencing on a CEQ8800 Sequencer (Beckman Coulter). Segregation in family members was performed for variants identified in the proband in heterozygous state in order to confirm that the variants were in trans (yes in column Seg in Table 1).
Statistical analysis
The relation between two class group variables was assessed by Fisher exact test.
Results
The 20 patients with Bardet-Biedl syndrome enrolled in this study were screened using a panel of 18 genes associated with the disease. We obtained the following results. No variants could be found in eight patients whose characteristics are described in Additional file 1: Table S3). We defined the causal variants in 12 patients (60%) (Table 1); 5 patients were confirmed to have a compound heterozygous variant in a BBS gene, while 7 patients where homozygous for the causative variant. In four cases the pathogenic variants were novel. We identified a novel compound heterozygous variant in BBS1 c.1285dup (p.(Arg429Profs*72); a likely pathogenic novel variant affecting the conserved residue 354 in the functional domain of BBS2 (c.1062C > G; p.(Asn354Lys)); a pathogenic new homozygous nucleotide change in BBS7 that leads to a stop codon in position 255, c.763A > T, and a likely pathogenic homozygous substitution c.1235G > T in BBS6, leading to the change p.(Cys412Phe). The novel variants were scored as pathogenic or likely pathogenic according to Richards et al. classification [18]. Four of the 12 patients in which the causative gene had been identified, also had potentially pathogenic variants in additional BBS genes suggesting oligogenic inheritance and a possible modifier effect. Three out of 4 patients presented variants in two additional genes, while patient #6 presented a variant with unknown significance (VUS) in BBS1 in addition to the “principal variants” in BBS12. Patient #4 presented “principal mutations” in BBS2 and additional functional polymorphism in BBS12 and a variant with unknown significance in INPP5; patient #10 presented additionally to the principal variant in BBS1, variants in BBS10 and BBS14, the first likely benign and the second with unknown significance; patients #11 with compound heterozygous variants in BBS4, presented additionally VUS in BBS8 and BBS2. Unlikely, we cannot document any influence on the phenotype severity in patient carrying additional variants in heterozygous state due to the small cohort of patients analysed and to the fact that all the variants were missense and with an uncertain role in disease manifestation.
In our cohort, we had a prevalence of patients harbouring genetic variants in BBS10 (N = 4), two patients with variants in BBS2 and two in BBS12. The remaining four patients had their causative pathogenic variants in each of the following genes: BBS1, BBS4, BBS6, BBS7.
The clinical characteristics of positive patients are reported in Table 2. Patients were predominantly males, and all were clinically diagnosed during childhood or adolescence (median age 21 months). All patients with a molecular diagnosis had polydactyly of one or both hands or feet, in most cases hexadactyly of feet as well as hands (N = 6) (Table 2 and Additional file 1: Table S2). All patients had cone-rod dystrophy / retinitis pigmentosa (CRD/RP), except one who had myopia and cataract (Additional file 1: Table S2), highlighting the fact that clinical diagnosis of BBS is often made when patients manifest ocular involvement. Other frequent clinical characteristics in our cohort were intellectual disability, hypogonadism, obesity and renal abnormalities (Additional file 1: Table S2). The cohort of negative patients was not statistically significantly different from the cohort of molecularly resolved patients (Additional file 1: Table S2).
Discussion
Bardet-Biedl syndrome, a ciliopathy with autosomal recessive/oligogenic inheritance shows high clinical variability and genetic heterogeneity. Although the postaxial polydactyly is evident at birth, clinical diagnosis is usually made when patients complain of visual distress. Genetic testing can help to confirm the diagnosis and enable prompt and effective clinical management. The molecular characterization of patients in addition can help to predict a grade of severity and to identify carrier’s family members with potential benefits of counselling. The targeted sequencing also has the advantage of allowing the identification of modifiers or genes with an epistatic effect that can shed light on phenotypic variability between and within families. Furthermore the molecular diagnosis can be fundamental for personalized medicine, aiming at the identification of patients potentially suitable for enrolment in any appropriate clinical trial that may become available in the future.
In this direction, recent advances in gene therapy and personalized medicine have enabled significant advances in the development of potential therapies for BBS patients, although the number of genes involved and the variety of mutations identified in patients, often in the same family, has increased the challenge faced by researchers [29].
Our study was performed to increase the BBS mutation spectrum in a cohort of 20 unrelated Italian patients with BBS. Genetic diagnosis was obtained in 12/20 patients using a NGS targeted approach analysing simultaneously the 18 most frequently mutated genes associated with BBS to increase the detection rate and the understanding of the BBS phenotype through discovery of additional mutations in BBS genes that could explain differences in phenotype severity.
The study revealed BBS10 gene mutations in a majority of our cohort (33%), in accordance with the percentages already reported in the literature [4]. Interestingly, we found just one patient with variants in BBS1, the most frequently detected gene in BBS patients [4, 30,31,32].
We identified a novel variant in BBS1 patient #10 c.1285dup (p.(Arg429Profs*72)) defined as pathogenic that segregates with phenotype together with c.46A > T (p.(Ser16Cys), defined as likely pathogenic.
A new pathogenic variant in BBS2 affecting a conserved residue in the functional domain of BBsome protein (c.1062C > G; p.(Asn354Lys)) was found in compound heterozygous state in patient #1 together with the known pathogenic variant p.(Arg339*). A new homozygous nucleotide change in BBS7 that leads to a stop codon in position 255, c.763A > T, was identified in patient #3. BBS1, BBS2 and BBS7 share a partially overlapping portion of a functional domain, mutation of which results in the same disease phenotype [30]. New pathogenic variants of BBS2 and BBS7 lie in this portion. The variant in BBS7 is noteworthy, since very few Bardet-Biedl cases are reported in the literature. Indeed, only 35 variants [33] in this gene are listed in the Human Gene Mutation Database (HGMD, https://portal.biobase-international.com/cgi-bin/portal/login.cgi). A homozygous substitution c.1235G > T in BBS6, leading to p.(Cys412Phe), was also identified in an affected sibling of proband #12. Interestingly, the clinical severity of the phenotypes of the two siblings was different (Table 2), suggesting that these variants show intrafamilial variable expressivity or that the patient’s genetic background strongly influences phenotype.
Interestingly, four of the twelve patients in which the causative gene had been identified, also had additional variants in BBS genes suggesting oligogenic inheritance and a possible modifier effect. However, we cannot document any influence on the phenotype severity in patient carrying additional variants in heterozygous state due to the small cohort of patients analysed and to the fact that all the variants were missense and with an uncertain role in disease manifestation. Our results are concordant with the results obtained also by other authors: the impact of the additional heterozygous variant remains elusive since it is very difficult to determine the contribution of the third allele to the phenotype. Available studies compare different kind of mutations, with different impact on the protein in small cohorts with high genetic heterogeneity [32, 34]. Moreover, in our cohort, we do not present family members with difference in the genetic background that could help shed light on the triallelic inheritance hypothesis [30].
In order to explore the function of different genes involved in BBS we looked for possible correlations in our cohort dividing patients with a molecular diagnosis with variants in BBSome genes or with variants in BBSome chaperonin genes. The clinical phenotype spectrum is wide (different type of mutations on different genes) and we did not observe any correlation between characteristic of patients probably due to the small number of patients analysed. This limits the possibility to highlight genotype-phenotype correlation with a statistical significance. Previous study have not identified any correlation between individual genotypes and phenotype [35, 36]. Interestingly, Billingsley et al. stated that patients with mutations in BBS10 or 12 had a similar phenotype, supporting our sorting of patients [33].
Characteristics such as obesity and intellectual impairment or retinal degeneration affected patients with mutations in genes of the BBSome or coding for BBSome chaperonin with the same frequencies. Hypogonadism (manifesting as genital anomalies in females and small penis buried in adipose tissue with undescended testes in males [11]) was more frequent in patients with variants in BBSome protein, whereas renal abnormalities were mostly present in patients with variations in BBSome chaperonin genes [12] (Table 3). Patients with renal anomalies should therefore be screened mainly in BBS10, BBS12, BBS6 and those with hypogonadism for variants in BBSome genes. Clinicians should also closely monitor patients harbouring mutations in BBS10, BBS12, BBS6 to favour early detection of those with renal anomalies, at risk of kidney failure and sudden death.
In our cohort, hexadactyly was found in all patients with a molecular diagnosis. Confirming the results of Beales et al., polydactyly of the toes was more common than that of the fingers in our cohort (11vs9), and both feet more often had polydactyly than both hands (90% vs 66%) [13].
Conclusion
In conclusion, our results demonstrate that NGS panels are a fast and effective way of obtaining high diagnostic yields in diseases, such as BBS, caused by mutations in many genes. They also provide information on other mutant genes in addition to the causative one. Genetic testing can provide insights into the pathways involved in the disease and aid the development of targeted therapy, which needs to begin with the first eye symptoms, before extensive photoreceptor damage. When feasible, NGS should be considered the elective method of genetic testing to confirm any hypothesis of tri-allelic inheritance. Our results are further evidence that BBS is extremely heterogeneous: by describing four new variants we extend the mutational spectrum of known BBS genes and contribute information on genotype-phenotype correlations. Finally, the fact that 40% of our patients did not have any mutation in the 18 known screened genes, leads us to the consideration that although being the best available approach, targeted NGS do not allow the detection of (i) deeply intronic variants that could have an effect either on the splicing or expression of the genes; (ii) large gene rearrangements nor (iv) the analysis of additional responsible loci yet to be identified. In addition given the fact that there is a significant overlap between clinical sign and mutated genes within BBS and other ciliopathies such as McKusick-Kaufman syndrome (MKKS; OMIM 604896), Alstrom syndrome (ALMS; OMIM 203800), Meckel-Gruber syndrome (MKS; OMIM 249000, 603294, 607361, 611134), and Joubert syndrome (JBTS; OMIM 213300, 608,091, 608629, 609583, 610688, 611560, 612291, 612285, 300804) we cannot exclude that our negative patients presented variants in genes more associated to the other syndromes and that were not sequenced.
Availability of data and materials
The novel variant identified during the current study are available in the Clinvar repository (SUB4899621).
Abbreviations
- BBS:
-
Bardet-Biedl syndrome
- CRD:
-
Cone-rod dystrophy.
- del:
-
Deletion
- dup:
-
Duplication
- ex:
-
Exon
- F:
-
Female
- fSNP:
-
Functional single nucleotide polymorphism
- het:
-
Heterozygous
- homo:
-
Homozygous
- ins:
-
Insertion
- int:
-
Intron
- LB:
-
Likely benign
- LP:
-
Likely pathogenic
- M:
-
Male
- MAF:
-
Minor allele frequency
- NGS:
-
Next generation sequencing
- P:
-
Pathogenic
- Ref:
-
References
- RP:
-
Retinitis pigmentosa
- RS:
-
dbSNP accession number
- seg:
-
Segregation performed
- VUS:
-
Variant unknown significance
References
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results. J Med Genet. 1999;36(6):437–46.
Riise R. The cause of death in Laurence-Moon-Bardet-Biedl syndrome. Acta Ophthalmol Scand Suppl. 1996;(219):45–7. PubMed PMID: 8741118.
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, et al. A Core complex of BBS proteins cooperates with the GTPase Rab8 to promote Ciliary membrane biogenesis. Cell. 2007;129(6):1201–13.
Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, Badano JL, et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet. 2006;38(5):521–4.
Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. Review. PubMed PMID: 17009929
Wheway G, Nazlamova L, Hancock JT. Signaling through the Primary Cilium. Front Cell Dev Biol. 2018;6:8. https://doi.org/10.3389/fcell.2018.00008. eCollection 2018. Review. PubMed PMID: 29473038; PubMed Central PMCID: PMC5809511.
Beales PL, Badano JL, Ross AJ, Ansley SJ, Hoskins BE, Kirsten B, et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl Syndrome. Am J Hum Genet. 2003;72(5):1187–99.
Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, Lewis RA, et al. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439(7074):326–30.
Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum Mol Genet. 2004;13(90001):65R–71.
Riise R, Andréasson S, Borgström MK, Wright AF, Tommerup N, Rosenberg T, et al. Intrafamilial variation of the phenotype in Bardet-Biedl syndrome. Br J Ophthalmol. 1997;81(5):378–85.
Priya S, Nampoothiri S, Sen P, Sripriya S. Bardet–Biedl syndrome: genetics, molecular pathophysiology, and disease management. Indian J Ophthalmol. 2016;64(9):620.
Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, et al. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol. 2011;6(1):22–9.
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–46.
Shaheen R, Szymanska K, Basu B, Patel N, Ewida N, Faqeih E, et al. Characterizing the morbid genome of ciliopathies. Genome Biol. 2016;17(1):242.
Muller J, Stoetzel C, Vincent MC, Leitch CC, Laurier V, Danse JM, et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet. 2010;127(5):583–93.
Nishimura DY, Searby CC, Carmi R, Elbedour K, Van Maldergem L, Fulton AB, et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2). Hum Mol Genet. 2001;10(8):865–74.
Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, et al. The human splicing code reveals new insights into the genetic determinants of disease. Science (80- ). 2015;347(6218):1254806.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23.
Zaghloul NA, Liu Y, Gerdes JM, Gascue C, Oh EC, Leitch CC, et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc Natl Acad Sci U S A. 2010;107(23):10602–7.
Sathya Priya C, Sen P, Umashankar V, Gupta N, Kabra M, Kumaramanickavel G, et al. Mutation spectrum in BBS genes guided by homozygosity mapping in an Indian cohort. Clin Genet. 2015;87(2):161–6.
Lindstrand A, Davis EE, Carvalho CMB, Pehlivan D, Willer JR, Tsai I-C, et al. Recurrent CNVs and SNVs at the NPHP1 locus contribute pathogenic alleles to Bardet-Biedl syndrome. Am J Hum Genet. 2014;94(5):745–54.
Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, Vicaire S, et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet. 2007;80(1):1–11.
Esposito G, Testa F, Zacchia M, Crispo AA, Di Iorio V, Capolongo G, et al. Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: identification of eleven novel pathogenic sequence variants. BMC Med Genet. 2017;18(1):10.
Janssen S, Ramaswami G, Davis EE, Hurd T, Airik R, Kasanuki JM, et al. Mutation analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals. Hum Genet. 2011;129(1):79–90.
Hjortshøj TD, Grønskov K, Philp AR, Nishimura DY, Riise R, Sheffield VC, et al. Bardet-Biedl syndrome in Denmark--report of 13 novel sequence variations in six genes. Hum Mutat. 2010;31(4):429–36.
Wiszniewski W, Lewis RA, Stockton DW, Peng J, Mardon G, Chen R, et al. Potential involvement of more than one locus in trait manifestation for individuals with Leber congenital amaurosis. Hum Genet. 2011;129(3):319–27.
Knopp C, Rudnik-Schöneborn S, Eggermann T, Bergmann C, Begemann M, Schoner K, et al. Syndromic ciliopathies: from single gene to multi gene analysis by SNP arrays and next generation sequencing. Mol Cell Probes. 2015;29(5):299–307.
Katsanis N, Eichers ER, Ansley SJ, Lewis RA, Kayserili H, Hoskins BE, et al. BBS4 is a minor contributor to Bardet-Biedl Syndrome and May also participate in Triallelic inheritance. Am J Hum Genet. 2002;71(1):22–9.
Forsythe E, Kenny J, Bacchelli C, Beales PL. Managing Bardet-Biedl Syndrome-now and in the future. Front Pediatr. 2018;6:23.
Badano JL, Ansley SJ, Leitch CC, Lewis RA, Lupski JR, Katsanis N. Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet. 2003;72(3):650–8.
M’hamdi O, Redin C, Stoetzel C, Ouertani I, Chaabouni M, Maazoul F, et al. Clinical and genetic characterization of Bardet-Biedl syndrome in Tunisia: defining a strategy for molecular diagnosis. Clin Genet. 2014;85(2):172–7 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23432027. [cited 2019 Mar 27].
Suspitsin EN, Imyanitov EN. Bardet-Biedl Syndrome. Mol Syndromol. 2016;7(2):62–71 Available from: https://www.karger.com/Article/FullText/445491. [cited 2019 Mar 27].
Billingsley G, Bin J, Fieggen KJ, Duncan JL, Gerth C, Ogata K, et al. Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. J Med Genet. 2010;47(7):453–63 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20472660. [cited 2019 Mar 29].
Khan SA, Muhammad N, Khan MA, Kamal A, Rehman ZU, Khan S. Genetics of human Bardet-Biedl syndrome, an updates. Clin Genet. 2016;90(1):3–15 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/cge.12737. [cited 2019 Mar 27].
Deveault C, Billingsley G, Duncan JL, Bin J, Theal R, Vincent A, et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum Mutat. 2011;32(6):610–9 Available from: http://www.ncbi.nlm.nih.gov/pubmed/21344540. [cited 2019 Mar 27].
Daniels AB, Sandberg MA, Chen J, Weigel-DiFranco C, Fielding Hejtmancic J, Berson EL. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130(7):901–7 Available from: https://jamanetwork.com/journals/jamaophthalmology/fullarticle/1149517. [cited 2019 Mar 27].
Acknowledgements
We are grateful to all the patients and their families, for their invaluable contributions. We would like to thank Helen Ampt for revising the manuscript and also all the lab member of the MAGI’s group.
Funding
We thank Provincia Autonoma di Bolzano for contributing to the funding of this project (n. 116/2016). The contribution by Fondazione Bietti in this paper was supported by the Ministry of Health and Fondazione Roma.
Author information
Authors and Affiliations
Contributions
EM designed the research, performed experiment, analysed the results and wrote the paper; SP, GG PEM performed experiment and revised paper; FD, AA, ZL, BF, LC, GI, AP,LB, MM, BM, LB, LR analysed the clinical data and revised the paper; MB designed the research and revised the paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study is a retrospective case series description that does not require ethics committee approval. Written, informed consent was obtained prior to their inclusion in this study. The research adhered to the tenets of the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. List of genes in the NGS panel. Table S2. Distribution of BBS diagnostic criteria in patients with molecular diagnosis. Table S3. Clinical manifestations of Bardet-Biedl syndrome patients with unresolved genotype. Abbreviations: M, male; F, female; yrs., years; mo, months; RP retinitis pigmentosa; CRD, cone-rod dystrophy; HM, high myopia; O, obese; OW, overweight; N/K, not known. (DOCX 28 kb)
Additional file 2:
Mutation analysis. The DNA probe set was designed using specific Illumina DesignStudio online tool (https://designstudio.illumina.com/). (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Manara, E., Paolacci, S., D’Esposito, F. et al. Mutation profile of BBS genes in patients with Bardet–Biedl syndrome: an Italian study. Ital J Pediatr 45, 72 (2019). https://doi.org/10.1186/s13052-019-0659-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-019-0659-1