
Abstract
Background
KLK10 exon 3 hypermethylation correlated to tumor-specific lack of KLK10 expression in cancer cell lines and primary tumors. In the present study we investigate the possible role of KLK10 exon 3 methylation in ovarian tumor diagnosis and prognosis.
Results
Qualitative methylation-specific PCR (MSP) results did not show statistically significant differences in patient group samples (normal and tumor) where all samples were positive only for the unmethylated-specific PCR except for two malignant samples that were either doubly positive (serous carcinoma) or doubly negative (Sertoli-Leydig cell tumor) for the two MSP tests. However, KLK10 exon 3 unmethylated PCR product concentration (ng/μl) showed statistically significant differences in benign and malignant patient group samples; mean ± SD (n): tumor: 0.077 ± 0.035 (14) and 0.047 ± 0.021 (15), respectively, p-value = 0.011; and normal: 0.094 ± 0.039 (7) and 0.046 ± 0.027 (6), respectively, p-value = 0.031. Moreover, ROC curve analysis of KLK10 exon 3 unmethylated PCR product concentration in overall patient group samples showed good diagnostic ability (AUC = 0.778; p-value = 0.002). Patient survival (living and died) showed statistically significant difference according to preoperative serum CA125 concentration (U/ml); median (n): 101.25 (10) and 1252 (5), respectively, p-value = 0.037, but not KLK10 exon 3 unmethylated PCR product concentration (ng/μl) in overall malignant patient samples; mean ± SD (n): 0.042 ± 0.015 (14) and 0.055 ± 0.032 (7), p-value = 0.228.
Conclusion
To the best of our knowledge, this is the first report on KLK10 exon 3 unmethylated PCR product concentration as potential early epigenetic diagnostic marker in primary ovarian tumors. Taken into account the limitations in our study (small sample size and semi-quantitative PCR product analysis) further studies are strongly recommended.
Similar content being viewed by others

Background
DNA methylation is one of the well-studied epigenetic modifications in DNA/chromatin metabolism. It is a dynamic process and involves the reversible and heritable methylation of the 5′ carbon of cytosine residues to yield 5-methylcytosine (5-MC) [1]. The reaction belongs to one carbon metabolism where DNA methyltransferases (DNMT1, DNMT3a and DNMT3b) are the biocatalysts and S-adenosylmethionine (SAM) is the methyl-donor [2]. Besides having important physiological roles in cell differentiation, development and gene regulation [3], DNA methylation can provide clues to other physiological processes, e.g. cell and tissue aging [4] and establishment of memory [5].
In DNA the cytosine residues occur either in frequencies that are far less than expected or in CpG-rich short stretches (0.5–4 kbp) in gene promoters and other regulatory regions known collectively as CpG islands [6]. In the CpG context the two cytosines on the opposing DNA strands are usually symmetrical as for their methylation status, i.e. both are either unmethylated or methylated [7]. The effect of DNA methylation on gene regulation may differ according to the context in which it occurs; however, in CpG-rich gene promoters it is well known to share in gene silencing [8]. Deregulated gene methylation was implicated in several diseases including cancer [9]. Nonetheless, aberrant gene methylation in cancer can be a promising diagnostic and prognostic target in tumor and naked DNA samples; e.g. in lung cancer [10].
The kallikrein-related peptidase 10 gene (KLK10) is one of 15 members in a serine protease gene subfamily located in tandem on chromosome 19q13.3–13.4 [11]. The gene product (KLK10) is a secreted protein found in normal human mammary epithelial cells (MECs) but downregulated or absent in the majority of human breast cancer cell lines [12]. KLK10 has a wide tissue expression [13] and is regulated by mechanisms that include steroid hormones [14] and micRNA [15]. The gene was reported as candidate tumor suppressor in some cancer types, e.g. in prostate cancer cells [16]; however, it showed contrasting expression profiles in different cancers, e.g. underexpressed in breast [17], testis [18] and prostate [19], and overexpressed in ovary [20], colon [21], and pancreas [22].
Liu and colleagues did not find mutation in KLK10 gene in different cancer types [12]. Indeed, tumor-specific lack of KLK10 expression correlated to KLK10 exon 3 hypermethylation in a majority of cell lines and in primary breast cancer [23]. The rationale behind such correlation may be understood as KLK10 exon 3 methylation was shown to perfectly follow that of KLK10 gene promoter [24]. Results of previous works were consistent about the confinement of KLK10 exon 3 hypermethylation in malignant samples; but not normal ones, in cell lines [23,24,25] and in primary tumors [23,24,25,26]. However, in some sample sets KLK10 expression did not show simple correlation with KLK10 exon 3 methylation [23,24,25,26]. In the present study, KLK10 exon 3 methylation is assessed for its possible role in the biology, diagnosis and/or prognosis of ovarian tumors.
Methods
Patients and samples
This is a further study for our previous work on serum KLK6 and 10 in ovarian cancer patients [27]. The protocol of the present study was approved by the Ethics and Research Committee of Ain Shams University.
Patients’ demographic data and preoperative serum marker levels (CA125, KLK10 and KLK6) were taken from our previous data as mentioned above. Patients or their relatives were contacted from October 2012 to January 2013 to collect patient follow-up data and have their written informed consent.
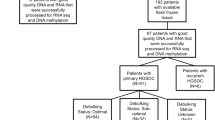
The studied samples were archival FFPE-ovarian tissue samples that are available at the Gynecologic Oncology Unit, Ain Shams University Maternity Hospital. The included samples were tumors and their contralateral normal ovarian samples (for some cases), while non-neoplastic ovarian masses, i.e. inflammation and endometriosis, were excluded.
Experimental protocol
Using histopathology microtome (microTec® cut 4050, UK) 10 to 15 tissue sections, each of ~ 2 × 2 cm in diameter and 1–2 μm thickness were cut and put in clean and autoclaved 1.5 ml microcentrifuge tube, one day before DNA extraction. An H&E stained tissue section was prepared and examined microscopically for each sample.
DNA extraction
Commercially available QIAamp DNA FFPE Tissue Kit (Qiagen) was used. The steps of the experiment were done according to the product insert provided by the manufacturer. Xylol (Sigma-Aldrich, Germany) was used to dissolve the paraffin and 98–100% ethanol (Sigma-Aldrich, Germany) was used to remove the xylol and to reconstitute the buffers. The concentration of the extracted pure DNA was measured spectrophotometrically (Ultraspec® 1000, Amersham Pharmacia Biotech, Cambridge, England) at 260 nm and the DNA samples were stored at − 80 °C until the time of DNA sodium bisulfite treatment.
Sodium bisulfite treatment of DNA
One μg of extracted DNA was treated by sodium bisulfite using commercially available EpiTect Bisulfite Kit (Qiagen). The steps of the experiment and the thermal cycler program were done according to the product insert given by the manufacturer for FFPE-samples. The sodium bisulfite converted DNA samples were kept at − 80 °C until the time of MSP experiment.
MSP for KLK10 exon 3
Commercially available HotStarTaq Master Mix kit (Qiagen) was used. Methylated- and unmethylated-specific PCRs were run in parallel in separate PCR tubes where each tube contained either methylated- or unmethylated-specific primer pair, respectively. The two primer pairs were provided by Invitrogen (USA), as described elsewhere [23]. Each PCR reaction contained ~ 0.5 μg sodium bisulfite-treated DNA sample. The concentrations of the PCR reaction components were made according to the manufacturer’s instructions in the product insert. A methylation positive control MSP was done using 2 μg (1 μg for each MSP test type) fully methylated DNA (EpiTect® Control DNA (human), methylated and bisulfite converted (100), Qiagen). The thermal cycler program was as follows: 1 cycle of 95 °C for 15 min; 35 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min; and 1 cycle of 72 °C for 10 min.
Capillary gel electrophoresis
PCR products were assessed by capillary gel electrophoresis using QIAxcel system (Qiagen). A QIAxcel kit (QIAxcel DNA High Resolution Kit (1200)) was used and the experiment steps were done according to the manufacturer’s instructions in the product insert. Determination of DNA fragment size and concentration was done using the BioCalculator software provided with the QIAxcel (Qiagen).
Statistical analysis
Statistical analysis of data was done using SPSS (version 15.0 for Windows). p < 0.05 was considered the cutoff value for significance. Chi-square test (χ2) was used to test the association of categorical data. Parametric or non-parametric tests were used to compare mean ± SD (Student t-test or ANOVA test) or median and interquartile range (Mann-Whitney test or Kruskal Wallis test) in two or more populations, respectively, involving independent samples. Spearman’s test was used to evaluate correlations between continuous variables. A receiver operating characteristic (ROC) curve was used to illustrate the diagnostic properties of a test on a numerical scale. Marker’s diagnostic parameters were calculated as follows: sensitivity = true positive tests/ all positive samples by gold standard test; specificity = true negative tests/ all negative samples by gold standard test; positive predictive value (PPV) = true positive tests/ all positive tests; negative predictive value (NPV) = true negative tests/ all negative tests; and accuracy = true positive and true negative tests/all positive and all negative tests.
Results
Patients and samples
This study included 14 benign and 16 malignant ovarian tumor patients. Patient clinicopthological data are shown in Table 1. There were no statistically significant differences of patient age mean ± SD according to disease grade (1, 2 and 3): mean ± SD (n): 36.8 ± 11.2 (5), 48.0 ± 19.6 (5) and 46.5 ± 11.0 (6), respectively; p-value = 0.422, or disease stage (I/II and III/IV): mean ± SD (n): 42.5 ± 16.4 (9) and 45.7 ± 11.7 (7), respectively; p-value = 0.675.
Each patient participated by one tumor sample. In addition, samples from contralateral normal ovaries of some patients in benign and malignant patient groups were included as normal controls (n = 7 and 6, respectively). Table 2 shows the pathological characteristics of tumor samples in the two patient groups. Histopathological H&E examination of all samples showed good quality of the tissue samples and confirmed the previously known diagnoses.
MSP
In the overall there were 42 unmethylated-specific positive PCR tests and only one methylated-specific positive PCR test. The unmethylated PCR band size was as follows: min.-max.: 101.30–274.00 bp; median: 123.45 bp and mean ± SD: 126.25 ± 23.61 bp, whereas the methylated band was 134.5 bp. The methylation positive control sample was positive for the methylated-specific PCR (band size: 129.0 bp) but negative for unmethylated-specific PCR.
Qualitative results of MSP did not show statistically significant difference in the two patient groups for both tumor and normal samples (Table 3). However, comparison of KLK10 exon 3 unmethylated PCR product concentration mean ± SD (ng/μl) showed statistically significant differences in benign (n = 14), malignant (n = 15) and overall normal (n = 13) samples; mean ± SD: 0.077 ± 0.035, 0.047 ± 0.021, and 0.072 ± 0.041, respectively; p-value = 0.046. Moreover, KLK10 exon 3 unmethylated PCR product concentration mean ± SD (ng/μl) showed statistically significant differences in tumor and normal samples in the two patient groups (Fig. 1).

KLK10 exon 3 unmethylated PCR product concentration (ng/μl) mean ± SD in patient group samples. Number of samples in benign and malignant patient groups were: normal sample: 7 and 6, respectively, and tumor sample: 14 and 15, respectively
Spearman’s correlation of KLK10 exon 3 unmethylated PCR product concentration (ng/μl) showed statistically significant positive correlation with patient age (ρ = 0.398; p-value = 0.009) and statistically significant negative correlation with serum KLK10 (ρ = − 0.300; p-value = 0.054) but not KLK6 (ρ = − 0.114; p-value = 0.471) or CA125 (ρ = − 0.214; p-value = 0.197). Comparisons of KLK10 exon 3 unmethylated PCR product concentration mean ± SD (ng/μl) in overall samples and in sample pathology types in the two patient groups according to patient age and serum KLK10 are shown in Tables 4 and 5, respectively.
ROC curves
ROC curves for KLK10 exon 3 unmethylated PCR product concentration (ng/μl) and preoperative serum markers in the two patient groups are shown in Figs. 2a and b, respectively.

ROC curve analysis of KLK10 exon 3 unmethylated PCR product concentration (ng/μl) in overall patient group samples (a) and ROC curves of preoperative serum markers CA125, KLK10 and KLK6 in patient groups (b). Markers’ diagnostic profiles were as follows: 
Combination of KLK10 exon 3 unmethylated PCR product concentration and serum marker showed enhanced combined marker sensitivity with all 3 markers and better specificity with CA125 but not KLK10 or KLK6. Combined marker profile of KLK10 exon 3 unmethylated PCR product concentration with serum marker was as follows; CA125: sensitivity: 100%, specificity: 94.4%, PPV: 68.9%, NPV: 73.9% and accuracy: 71.1%; KLK10: sensitivity: 90.4%, specificity: 85.7%, PPV: 67.8%, NPV: 62% and accuracy: 64.9%; and KLK6: sensitivity: 85.7%, specificity: 71.4%, PPV: 60%, NPV: 60% and accuracy: 60%.
Patients’ survival
Five-year patient follow-up data were available between 2007 and 2012. Five patients died within the first two years of the follow-up period. In the living 11 patients there were 9 disease-free and 2 relapsed. Comparisons of patient clinicopathological parameters according to patient survival (living and died) showed statistically significant differences in preoperative serum CA125 concentration (U/ml); median (n): 101.25 (10) and 1252 (5), respectively; p-value = 0.037, and disease stage (I/II and III/IV): late stage was more frequent in died patients compared to the living (4 vs. 1, respectively); p-value = 0.049. The p-values for other clinicopathological parameters were as follows: patient age mean ± SD: 0.355; serum KLK10 (ng/ml) mean ± SD: 0.798; serum KLK6 (ng/ml) mean ± SD: 0.278; ascites (absent and present): 0.106; disease grade (1, 2, and 3): 0.622; pathology subgroup (epithelial, nonepithelial and metastatic): 0.402; and epithelial pathology subtype (serous, mucinous, endometrioid and clear cell carcinoma): 0.735.
Comparison of KLK10 exon 3 unmethylated PCR product concentration (ng/μl) mean ± SD according to patient survival (living and died) did not show statistically significant differences in overall malignant patient samples: mean ± SD (n): 0.042 ± 0.015 (14) and 0.055 ± 0.032 (7), respectively; p-value = 0.228, normal samples: mean ± SD (n): 0.035 ± 0.010 (4) and 0.070 ± 0.042 (2), respectively; p-value = 0.153, or malignant samples: mean ± SD (n): 0.046 ± 0.017 (10) and 0.050 ± 0.030 (5), respectively; p = 0.748. However, the oddly methylated-specific PCR positive sample in our results was serous carcinoma of grade 3, stage III/IV and its unmethylated PCR product concentration was 0.06 ng/μl; the patient of which died one year after diagnosis and start of treatment.
Discussion
In the literature we could find only one study on KLK10 exon 3 methyaltion in primary ovarian tumors that was reported by Sidiropoulos and colleagues [25]. Our results agree with those in the previous work with some logic approximation due to differences in the used technique for gene methylation assessment (MSP vs. direct DNA sequencing, respectively) and the examined KLK10 expression parameter (serum KLK10 vs. cytosolic KLK10, respectively). The two studies showed KLK10 exon 3 methylation only in malignant samples (methylated-specific PCR positive samples: 1/16 and samples with partially methylated KLK10 exon 3: 6/7, respectively) but not in normal samples (methylated-specific PCR negative samples: 13/13 and samples with fully unmethylated KLK10 exon 3: 2/2, respectively). Additionally, we studied KLK10 exon 3 methylation in benign ovarian tumors – not included in the previous work – and they all were unmethylated (Table 3). Differences in the internal working of MSP and direct DNA sequencing would make comparison of frequencies of methylated samples in the two studies practically unfeasible (see later).
Moreover, as for the correlation between KLK10 gene expression and KLK10 exon 3 methylation the two studies showed similar results. In the previous work there was fair KLK10 expression in normal samples (fully unmethylated) and high expression in the partially methylated malignant samples (3 out of 6). In concordance to those results our samples showed negative correlation between serum KLK10 level (ng/ml) and KLK10 exon 3 unmethylated PCR product concentration (ng/μl) by Spearman’s correlation and by comparing the unmethylated PCR product concentration mean ± SD in overall samples according to KLK10 level (Table 5).
While our qualitative MSP results did not show statistically significant differences in patient group samples (Table 3), KLK10 exon 3 unmethylated PCR product concentration (ng/μl) mean ± SD showed statistically significant differences in normal and tumor samples in the two patient groups (Fig. 1). Because our MSP experiment conditions were almost constant for all PCR tests (including the operator, instruments and machines, kits and reagents, DNA template concentration, PCR program, simultaneous unmethylated- and methylated-specific PCRs, and random sample sets in 4 PCR runs done in 4 different days) those statistically significant differences of KLK10 exon 3 unmethylated PCR product concentration should be due to differences in the methylation patterns of samples rather than due to unfounded chance (see later).
Individual cells in the same tissue may show heterogeneity as regard their DNA methylation patterns [28]. Indeed, understanding that methylation pattern heterogeneity is fundamental in interpreting qualitative and quantitative DNA methyaltion data and also for appreciating and understanding several aspects of gene functions. Each cell contains two copies of each CpG site; one on each of the two homologous chromosomes, and, therefore, can have on its own 3 different methylation patterns for that particular CpG site: fully unmethylated, fully methylated or partially methylated. The possible methylation patterns of a number (n) of CpG sites are given mathematically as 2n. For example, when studying an average number of 12 CpGs by MSP, the possible methylation patterns in the DNA sample are 212 = 4096, which reflects possible immense methylation pattern heterogeneity when the cell can have any two of these patterns. However, in the context of KLK10 exon 3 in ovarian cancer the property of methylation pattern heterogeneity would be present almost exclusively in malignant samples but not in normal ones because in the previous work the malignant samples were partially methylated, whereas the normal samples were fully unmethylated [25].
In Fig. 3 we present a theoretical rationale for qualitative and quantitative MSP results based on possible methylation pattern heterogeneity in individual cells of a sample [28], the independency of the primer pairs in MSP from one another [23, 29] and previous DNA methylation results by MSP and direct DNA sequencing in cell lines and primary breast cancer [23]. We hypothesize that the existence of different partially methylated patterns in the sample would result in the stratification of the starting DNA template into template classes with varying MSP-expressivities according to the number of methylation-wise matching CpG sites with their corresponding nucleotides in the respective PCR primer. While some patterns would be more or less favorable for primer annealing (Fig. 3: elements C, and E-J), other patterns may not be at all (Fig. 3: element D). Differences of the proportions of those differentially MSP-expressible partially methylated patterns in the samples may be effected practically as differences in the unmethylated PCR product concentration. Given that those partially methylated patterns would occur only in malignant samples but not in normal – and probably also benign – samples (see above), such proportionate decreases in the unmethylated PCR product concentration would happen in malignant samples only.

The Figure shows a schematic representation of a theoretical rationale for MSP results based on previous works [23, 28, 29]. The DNA templates (long bars with 3′- > 5′ directionality designation) show blue and red dots representing unmethylated and methylated CpGs, respectively. The short blue and red bars represent unmethylated- and methylated-specific primer pairs, respectively. For simplicity, we show methylation patterns with uniform methylation status at the primer’s annealing site on DNA template (a-h) and only two with non-uniform methylation status (i and j). While fully unmethylated (a) and fully methylated (b) patterns provide unmethylated- and methylated-specific positive PCRs, respectively, partially methylated patterns (c-j) can have any of the four possible qualitative MSP results. MSP-inexpressible partially methylated pattern (d) may rationalize our doubly negative malignant sample (Sertoli-Leydig cell tumor), in addition to possible mutation as methylated CpGs are known hotspot for mutations [39, 40]
Our results showed statistically significant positive correlation between KLK10 exon 3 unmethylated PCR product concentration (ng/μl) and patient age by Spearman’s correlation and on comparing its mean ± SD in overall samples and in malignant samples according to age groups (Table 4). However, because there was no statistically significant difference of patient age in the two patient groups (Table 1), comparison of KLK10 exon 3 unmethylated PCR product concentration in patient group samples should be valid and justifiable.
KLK10 exon 3 methylation correlated to high disease grade (breast cancer [30]), late stage (lung cancer [26]) and bad patient prognosis (acute lymphoblastic leukemia [24]). Similarly, the only methylated-specific PCR positive sample in our results was serous carcinoma of high grade (3) and late stage (III/IV) whose patient died within one year after starting the treatment. However, in our results comparison of KLK10 exon 3 unmethylated PCR product concentration (ng/μl) according to disease grade, stage or patient survival did not show statistically significant differences that may be attributable to early implication of deregulated KLK10 exon 3 methylation in the disease or lack of significant information about site-specific CpG methylation in MSP data (see later).
The notion that information about the exact location of DNA methylation maybe crucial for correct clues about disease diagnosis, prognosis and gene function [31] would greatly restrain the translational impact of DNA methylation data by MSP. For example, lack of site-specific CpG methylation data (in MSP) and/or data about their pattern distribution in the sample (in MSP and direct DNA sequencing, respectively) may explain – at least in part – the inconsistent results, in sample subsets or sample types, about the downregulation of KLK10 through KLK10 exon 3 hypermethylation in previous works [23,24,25,26]. At the functional aspect, the reported antitumor role of KLK10 was demonstrated in vector-mediated transfection experiments in vitro and/or in tumor xenograft in mice using different cancer cell lines; e.g. breast [32], prostate [16] and gastric cell [33], including ovarian cell line [34]. Noteworthy is that overexpression of KLK10 was found in malignant sample subsets (tissues or sera) in cancer types in which KLK10 was shown experimentally as tumor suppressor; breast [35], prostate [19], gastric [36] and ovary [20, 27]. Taken together, the antitumor role of KLK10 may be understood only in relation to the context of the given study as regard type of tissue and the underlying pathophysiologic processes [37].
Indeed, the translational impact of DNA methylation data can vary greatly with the pros and cons of the used tools. While MSP studies only a few of CpGs without regard to their exact location, direct DNA sequencing can give site-specific CpG methylation data for all CpGs of interest as percent methylation in the sample. Unfortunately, both tools fall short of providing information about site-specific CpG methylation pattern distribution in sample. The more recently discovered next generation sequencing have the advantage of presenting site-specific DNA methylation data in absolute numbers and may tackle the problem of methylation pattern distribution in hand of sophisticated computational methods [38]. Nonetheless, considering possible dynamicity of DNA methylation as a mechanism for gene regulation [1] another dimension for DNA methylation pattern resolution may be necessary in order to spot and trace such spatiotemporal DNA methylation events rather than making statements based merely on single static snapshot image.
Conclusions
In summary, our results agreed with those in the previous work as for the exclusive occurrence of KLK10 exon 3 methylation in malignant but not in normal ovarian samples. Additionally, we assessed KLK10 exon 3 methylation in benign ovarian tumors (not included in the previous work) and found that they were unmethylated. Although our qualitative MSP results did not show statistically significant differences in patient group samples, semi-quantitative KLK10 exon 3 unmethylated PCR product concentration (ng/μl) showed statistically significant differences not only in tumor samples but also in otherwise histologically normal samples in benign and malignant patients. These findings would suggest, for the first time, KLK10 exon 3 unmethylated PCR product concentration as potential early epigenetic diagnostic marker in ovarian cancer. The diagnostic potential of KLK10 exon 3 unmethylated PCR product concentration in overall patient group samples was also evident in ROC curve analysis, and it could enhance the sensitivity of preoperative serum markers CA125, KLK10 and KLK6. Taking into account the limitations in the present study (small sample size and semi-quantitative assessment of PCR product concentration) further studies are needed before making a statement about the validity and reproducibility of these results. Moreover, we highlighted the translational limitations of DNA methylation data in MSP and direct DNA sequencing and addressed future challenges of the more recent next generation sequencing (NGS) to further enhance the translational impact of DNA methylation data.
Abbreviations
- 5-MC:
-
5-methylcytosine
- bp:
-
Base pair
- CA125:
-
Cancer antigen 125
- DNA:
-
Deoxyribonucleic acid
- DNMT1:
-
DNA methyltransferase 1
- DNMT3a:
-
DNA methyltransferase 3a
- DNMT3b:
-
DNA methyltransferase 3b
- FFPE:
-
Formalin-fixed paraffin-embedded
- H&E:
-
Hematoxylin and eosin
- KLK10:
-
Kallikrein-related peptidase 10
- KLK10 :
-
Kallikrein-related peptidase10 gene
- KLK6:
-
Kallikrein-related peptidase 6
- MECs:
-
Human mammary epithelial cells
- micRNA:
-
micro RNA
- MSP:
-
Methylation-specific PCR
- NEC:
-
Human normal epithelial cell line
- NGS:
-
Next generation sequencing
- NPV:
-
Negative predictive value
- PCR:
-
Polymerase chain reaction
- PPV:
-
Positive predictive value
- SAM:
-
S-adenosylmethionine
References
Wallace K, Grau MV, Levine AJ, Shen LL, Hamdan R, Chen XL, Gui JA, Haile RW, Barry EL, Ahnen D, McKeown-Eyssen G, Baron JA, Issa JPJ. Association between folate levels and CpG Island Hypermethylation in normal colorectal mucosa. Cancer Prev Res (Phila). 2010;3:1552–64.
Bird A, Tate P, Nan X, Campory J, Meehan R, Cross S, Tweedie S, Charlton J, Macleod D. Studies of DNA methyaltion in animals. J Cell Sci. 1995;Suppl 19:37–9.
Bird AP. Functions of DNA methylation in vertebrates. Cold Spring Harb Symp Quant Biol. 1993;58:281–5.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; https://doi.org/10.1186/gb-2013-14-10-r115.
Miller C, Sweatt J. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–69. https://doi.org/10.1016/j.neuron.2007.02.022.
Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. Mol Biol. 1987;196:261–82.
Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10(11):805–11.
Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–90.
Costello JF, Plass C. Methylation matters. J Med Genet. 2001;38(5):285–303.
Balgkouranidou I, Triantafyllou P, Theodoropoulos C, Tsaroucha EG, Kaklamanis L, Lianidou E. Prognsotic significance of DNA methylation markers in tissue and corresponding cell-free circulating DNA in plasma of NSCLC patients, vol. 2009. Denver: AACR 2009. 100st Annual Meeting of American Association of Cancer Research (AACR).
Yousef GM, Chang A, Scorilas A, Diamandis EP. Genomic organization of the human kallikrein gene family on chromosome 19q13.3-q13.4. Biochem Biophys Res Commun. 2000;276(1):125–33.
Liu XL, Wazer DE, Watanabe K, Band V. Identification of a novel serine protease-like gene, the expression of which is down-regulated during breast cancer progression. Cancer Res. 1996;56(14):3371–9.
Shaw JL, Diamandis EP. Distribution of 15 human kallikreins in tissues and biological fluids. Clin Chem. 2007;53(8):1423–32.
Luo LY, Grass L, Diamandis EP. Steroid hormone regulation of the human kallikrein 10 (KLK10) gene in cancer cell lines and functional characterization of the KLK10 gene promoter. Clin Chim Acta. 2003;337(1–2):115–26.
Chow TF, Crow M, Earle T, El-Said H, Diamandis EP, Yousef GM. Kallikreins as microRNA targets: an in silico and experimental-based analysis. Biol Chem. 2008;389(6):731–8.
Hu J, Lei H, Fei X, Liang S, Xu H, Qin D, Wang Y, Wu Y, Li B. NES1/KLK10 gene represses proliferation, enhances apoptosis and downregulates glucose metabolism of PC3 prostate cancer cells. Sci Rep. 2015; https://doi.org/10.1038/serp17426.
Dhar S, Bhargava R, Yunes M, Li B, Goyal J, Naber SP, Wazer DE, Band V. Analysis of normal epithelial cell specific-1 (NES1)/kallikrein 10 mRNA expression by in situ hybridization, a novel marker for breast cancer. Clin Cancer Res. 2001;7(11):3393–8.
Luo LY, Rajpert-De Meyts ER, Jung K, Diamandis EP. Expression of the normal epithelial cell-specific 1 (NES1; KLK10) candidate tumor suppressor gene in normal and malignant testicular tissue. Br J Cancer. 2001;85(2):220–4.
Petraki CD, Gregorakis AK, Papanastasiou PA, Karavana V, Luo LY, Diamandis EP. Immunohistochemical localization of human kalikreins 6, 10 and 13 in benign and malignant prostatic tissues. Prostate Cancer Prostatic Dis. 2003;6(3):223–7.
Shvartsman HS, Lu KH, Lee J, Lillie J, Deavers MT, Clifford S, Wolf JK, Mills GB, Bast RC Jr, Gershenson DM, Schmandt R. Overexpression of kallikrein 10 in epithelial ovarian carcinoma. Gynecol Oncol. 2003;90(1):44–50.
Talieri M, Alexopoulou DK, Scorilas A, Kypraios D, Arnogiannaki N, Devetzi M, Patsavela M, Xynopoulos D. Expression analysis and clinical evaluation of kallikrein-related peptidase 10 (KLK10) in colorectal cancer. Tumour Biol. 2011;32(4):737–44.
Rückert F, Hennig M, Petraki CD, Wehrum D, Distler M, Denz A, Schröder M, Dawelbait G, Kalthhoff H, Saeger HD, Diamandis EP, Pilarsky C, Grützmann R. Co-expression of KLK6 and KLK10 as prognostic factors for survival in pancreatic ductal adenocarcinoma. Br J Cancer. 2008;99(9):1484–92.
Li B, Goyal J, Dhar S, Dimri G, Evron E, Sukumar S, Wazer DE, Band V. CpG methylation as a basis for breast tumor-specific loss of NES1/KLK10 expression. Cancer Res. 2001;61(21):8014–21.
Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Barrios M, Andreu EJ, Prosper F, Heiniger A, Torres A. The normal epithelial cell-specific 1 (NES1) gene, a candidate tumor suppressor gene on chromosome 19q13.3-4, is downregulated by hypermethylation in acute lymphoblastic leukemia. Leukemia. 2004;18(2):362–5.
Sidiropoulos M, Pampalakis G, Sotiropoulo G, Katsaros D, Diamandis EP. Downregulation of human kallikrein 10 (KLK10/NES1) by CpG hypermethylation in breast, ovarian and prostate cancers. Tumour Biol. 2005;26(6):324–36.
Zhang Y, Song H, Miao Y, Wang R, Chen L. Frequent transcriptional inactivation of Kallikrein 10 gene by CpG island hypermethylation in non-small cell lung cancer. Cancer Sci. 2010;101(4):934–40.
El Sherbini MA, Sallam MM, Shaban EA, El-Shalakany AH. Diagnostic value of serum kallikrein-related peptidases 6 and 10 versus CA125 in ovarian cancer. Int J Gynecol Cancer. 2011;21(4):625–32.
Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for Folate’s role. Adv Nutr. 2012;3:21–38. https://doi.org/10.3945/an.111.000992.
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–6.
Kioulafa M, Kaklamanis L, Stathopoulos E, Mavroudis D, Georgoulias V, Lianidou ES. Kallikrein 10 (KLK10) methylation as a novel prognostic biomarker in early breast cancer. Ann Oncol. 2009;20(6):1020–5.
van Vlodrop IJH, Niessen HE, Derks S, Baldewijns MM, van Criekinge W, Herman JG, van Engeland M. Analysis of promoter CpgG island hypermethylation in cancer: location, location, location! Clin Cancer Res. 2011;17(13):4225–31.
Goyal J, Smith KM, Cowan JM, Wazer DE, Lee SW, Band V. The role for NES1 serine protease as a novel tumor suppressor. Cancer Res. 1998;58(21):4782–6.
Huang W, Tian X-L, Wu Y-L, Zhong J, Yu L-F, Hu SP, Li B. Suppression of gastric cancer growth by baculovirus vector-mediated transfer of normal epithelial cell specific-1 gene. World J Gastroenterol. 2008;14:5810–5.
Pépin D, Shao ZQ, Huppé G, Wakefield A, Chu CW, Sharif Z, Vanderhyden BC. Kallikreins 5, 6 and 10 differentially alter pathophysiology and overall survival in an ovarian cancer xenograft model. PLoS One. 2011;6(11):e26075. https://doi.org/10.1371/journal.pone.0026075.
Ewan King L, Li X, Cheikh Saad Bouh K, Pedneault M, Chu CW. Human kallikrein 10 ELISA development and validation in breast cancer sera. Clin Biochem. 2007;40(13–14):1057–62.
Jiao X, Lu H-J, Zhai M-M, Zhang D-P. Overexpression of kallikrein gene 10 is a biomarker for predicting poor prognosis in gastric cancer. World J Gastroenterol. 2013;19(48):9425–31.
Borgoño CA, Diamandis EP. The emerging roles of human tissue kallikreins in cancer. Nat Rev Cancer. 2004;4:876–90. https://doi.org/10.1038/nrc1474.
Xu H, Podolsky RH, Ryu D, Wang X, Su S, Shi H, George V. A method to detect differentially methylated loci with next-generation sequencing. Genet Epidemiol. 2012;37(4):377–82.
Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–5.
Rideout WM, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science. 1990;249(4974):1288–90.
Acknowledgments
We thank Dr. Mohamed Badr (PhD) from the Medical Research Center, Ain Shams Faculty of Medicine for helping in DNA extraction. We thank also Drs. Nashwa Khazragy (PhD, MD), Manar Mahmoud (PhD), Fatma Shehata (PhD) and Aisha Hussein (PhD) from the Oncology Diagnostic Unit, Biochemistry Department, Ain Shams Faculty of Medicine for helping in MSP experiments. Our thanks are due to Colors for Research laboratory in Cairo for conducting the capillary gel electrophoresis experiments. We are also much indebted to Drs. Hala El Sallaly (PhD, MD) and Sahar Ezz Elarab (PhD, MD) for helping in histopathology grading and in the statistical analysis of data, respectively.
Funding
This work was supported in part by cash money grant given once to PhD students (MAE) by The Academy of Scientific Research and Technology in Cairo. The funding body had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All data generated or analyzed during this study were included in this published article.
Author information
Authors and Affiliations
Contributions
MAE and AAM contributed equally to the conception and design of this work. MAE collected patient data and tissue samples and performed the experiments. ZAS performed and examined H&E sections of tissue samples. MAE interpreted the data and wrote the manuscript. AAM, MMS, EAS, ZAS and AHE revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol of the present study was approved by the Ethics and Research Committee of Ain Shams University. An informed written consent was taken from the living participants and relatives of died patients.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
El Sherbini, M.A., Mansour, A.A., Sallam, M.M. et al. KLK10 exon 3 unmethylated PCR product concentration: a new potential early diagnostic marker in ovarian cancer? - A pilot study. J Ovarian Res 11, 32 (2018). https://doi.org/10.1186/s13048-018-0407-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-018-0407-y