
Abstract
Cellular homeostasis requires the proper nuclear-cytoplasmic partitioning of large molecules, which is often deregulated in cancer. XPO1 is an export receptor responsible for the nuclear-cytoplasmic transport of hundreds of proteins and multiple RNA species. XPO1 is frequently overexpressed and/or mutated in human cancers and functions as an oncogenic driver. Suppression of XPO1-mediated nuclear export, therefore, presents a unique therapeutic strategy. In this review, we summarize the physiological functions of XPO1 as well as the development of various XPO1 inhibitors and provide an update on the recent clinical trials of the SINE compounds. We also discuss potential future research directions on the molecular function of XPO1 and the clinical application of XPO1 inhibitors.
Similar content being viewed by others

Background
Eukaryotic cells have well-separated nuclear and cytoplasmic compartments. Proper cellular functions require the exchange of large molecules through the nuclear pore complex (NPC). Small molecules can passively diffuse through the NPC, whereas the transport of larger cargoes, including RNAs and proteins, requires various transport receptors of the importin beta superfamily [1, 2]. XPO1 is a major transport receptor, responsible for exporting proteins and multiple RNA species. Originally named as CRM1 (chromosomal region maintenance 1), XPO1 was identified in Schizosaccharomyces pombe as a gene required for maintaining higher-order chromosome structure [3]. Subsequently, it was shown to function as a shuttling protein, mediating the nuclear export of proteins and mRNAs in Saccharomyces cerevisiae and renamed as XPO1 (exportin 1) [4].
Physiological functions of XPO1/CRM1
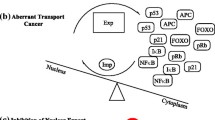
XPO1 is a nuclear export receptor with a pleiotropic role in transporting a plethora of proteins and RNA species, including rRNAs, snRNAs, mRNA, microRNAs, and tRNAs [5] (Fig. 1). XPO1 functions together with RAN GTPase, which provides the energy for transport and ensures the directionality of nuclear export [6]. In the nucleus, XPO1 binds to the nuclear export signal (NES) on its target proteins and to RAN in its active GTP-bound form (RAN-GTP). The complex is subsequently docked to NPC and passes through the nuclear membrane into the cytoplasm. Hydrolysis of RAN-GTP to RAN-GDP causes the disassembly of the complex and release of cargoes in the cytoplasm. The directionality of XPO1-mediated export is determined by the concentration gradient of RAN-GTP, which is predominantly confined to the nucleus [7] (Fig. 1). In addition to its role in nuclear-cytoplasmic transport during the interphase of cell cycle, XPO1/RAN regulates mitosis.

XPO1 mediates the nuclear export of hundreds of proteins and multiple RNA species
Protein export
XPO1 is involved in the export of nearly 220 proteins bearing NESs [8]. Among these proteins, several tumor suppressors, including p53, BRCA1/2, and p27, have been extensively studied. Nuclear export blockade of tumor suppressor proteins has been postulated as the primary mechanism of action (MOA) for XPO1 inhibitors [9, 10]. However, many known oncoproteins, such as SNAIL, cyclins, TERT/telomerase, SURVIVIN, DNA topoisomerases, c-ABL, and YAP1, are also exported by XPO1 [8, 11]. The indiscriminate export of tumor suppressors and oncogenes by XPO1 argues against nuclear retention of tumor suppressors as the major MOA for XPO1 inhibitors. Indeed, XPO1 inhibitors have been demonstrated to exhibit antitumor activities independent of the function of key tumor suppressor proteins, including RB, p53, and p21 [12,13,14]. The number of proteins exported by XPO1 may have been remarkably underestimated by earlier studies. A recent deep proteomic characterization of XPO1 protein cargoes has identified > 700 export substrates from S. cerevisiae, > 1000 from Xenopus oocytes, and > 1050 from human cells. The protein partitioning data suggest broad XPO1 functions in the regulation of vesicle coat-assembly, centrosomes, autophagy, peroxisome biogenesis, cytoskeleton, ribosome maturation, translation, and mRNA degradation [15]. This study concludes that XPO1-mediated protein export is general and promiscuous and that the impaired export of tumor suppressors may be one of the multiple potential mechanisms of action for XPO1 inhibitors.
RNA export
XPO1 has a major role in the nuclear export of multiple RNA species. First, XPO1 mediates the export of 40s and 60s ribonucleoprotein (RNP) complex in lieu of the naked ribosomal RNAs (rRNAs). Biogenesis of ribosomal subunits involves the synthesis of structural rRNAs and ribosomal proteins; their assembly into pre-ribosomal subunits in the nucleolus, export by XPO1; and further processing before gaining translational competency [16]. Second, XPO1 is critical for mRNA splicing by regulating the maturation of small nuclear RNAs (snRNAs). Following transcription in the nucleus, U snRNAs interact with the adaptor protein PHAX, RAN-GTP, and XPO1 to form an export-competent assembly. Exported U snRNAs are released in the cytoplasm, modified, and assembled into U snRNPs, before being shuttled back into the nucleus for further assembly into spliceosomes [17]. Third, XPO1 is involved in the export of other small non-coding RNAs, including microRNAs and tRNAs. microRNA and tRNA precursors are primarily exported by exportin 5 (XPO5) and exportin t (XPOT), respectively. However, XPO1 can mediate the alternative export of both microRNAs and tRNAs [18,19,20,21,22]. Fourth, XPO1 also exports mRNAs. mRNA is exported through either the bulk NXF1-mediated or the selective XPO1-mediated pathway [23, 24]. In particular, XPO1 and additional adaptor proteins with RNA binding properties, including LRPPRC, eIF4E, NXF3, and HuR, can preferentially export a subset of mRNAs encoding oncoproteins [25,26,27,28]. The diversity of the RNA species exported by XPO1 indicates that the inhibition of XPO1 may have a profound impact on different aspects of RNA metabolism.
Export-independent function
XPO1/RAN complex carries out nuclear export function during interphase, with an intact nuclear membrane. Equally important, however, is the export-independent function of XPO1/RAN during mitosis [29, 30]. XPO1 was originally identified as CRM1, with an essential role in regulating mitosis and chromosomal structure. XPO1 has been shown to localize to mitotic kinetochores [31] and is required for microtubule nucleation [32]. It is also present at centrosomes during cell cycle and may be involved in the recruitment of centrosomal scaffold proteins and assembly of mitotic spindles [33]. The transport-independent function is not limited to XPO1/RAN, as other members of the importin beta family and nucleoporins also play a role in regulating spindle assembly and kinetochore function [29, 30].
XPO1/CRM1 as an oncogenic driver and therapeutic target in cancer
XPO1 overexpression is a common feature among many human cancer types, including pancreatic, ovarian, glioma, lung, gastric, prostate, and colorectal cancers, and is associated with poor prognosis [34,35,36,37,38,39,40]. While amplified copy number may explain the XPO1 overexpression in some leukemia and lymphoma subtypes [41], for the majority of human cancers, the mechanism of XPO1 overexpression remains unknown. Genetic studies demonstrate a converse role for MYC and P53 in regulating XPO1 transcription [42]. The MYC overexpression and/or P53 loss of function observed in most human cancers are likely responsible for the widespread XPO1 overexpression. Interestingly, several XPO1 binding partners and/or adaptor proteins involved in nuclear export, namely RAN, HuR, eIF4E, LRPPRC, and NXF3, are also frequently overexpressed in human cancers and correlate with poor prognosis [24, 43,44,45,46,47], pointing to the aberrations of nuclear export machinery as a major hallmark of tumorigenesis. In addition to overexpression, XPO1 mutations have been found in 0.5–2.9% of solid and hematopoietic tumors, with the highest incidence in lymphomas [48]. A recent large-scale analysis of whole-exome and genome sequencing data from 42,793 patients has identified highly recurrent mutations of XPO1 (E571, R749, and D624) specifically in B cell malignancies. The E571K mutation is enriched in primary mediastinal B cell lymphoma (33%), classic Hodgkin lymphoma (14%), diffuse large B cell lymphoma (DLBCL) (2%), and chronic lymphocytic leukemia (CLL) (3%) [49]. Furthermore, XPO1E571K mutation has been shown to cooperate with MYC and BCL2 in promoting lymphomagenesis. These observations suggest that gain-of-function mutations convert XPO1 to an oncogenic driver, particularly in B cell malignancies. The exclusive presence of XPO1 mutations in B cell malignancies is largely unknown but is likely related to somatic hypermutations of immunoglobulin genes, where the error-prone polymerase η may introduce a high frequency of A > T and C > G transversions [50,51,52]. Due to frequent deregulation in cancers, XPO1 has been identified as a therapeutic target in many tumor types. Recent CRISPR library and RNAi screens have validated XPO1 as a therapeutic target in sarcoma, DLBCL, multiple myeloma, and KRAS-mutant lung cancer [9, 53,54,55]. Specific XPO1 inhibitors have been extensively tested and demonstrated efficacy in a broad range of cancer types in preclinical studies.
Development and preclinical study of XPO1/CRM1 inhibitors
The development of XPO1/CRM1 inhibitors dates back to early 1980s with the discovery of multiple classes of compounds and has been elegantly summarized in another review [56]. The first characterized XPO1 inhibitor, leptomycin B, was isolated from a strain of Streptomyces as an antifungal agent [57, 58]. Shortly following isolation and purification, its antitumor efficacy was examined in murine transplantation tumor models and demonstrated survival benefits [59]. However, a phase 1 trial of leptomycin B carried out in a small cohort of patients with various advanced cancers demonstrated marked malaise and anorexia, with no partial response [60]. Further clinical development of leptomycin B was therefore halted following the failed trial. XPO1/CRM1 was subsequently identified as the molecular target of leptomycin B with covalent binding at Cys528 [61]. Leptomycin B analogs, including ratjadones, anguinomycins, and KOS2464, also covalently bind to XPO1/CRM1 [62,63,64]. In particular, the semisynthetic KOS2464 has demonstrated significant therapeutic efficacy in several solid and hematopoietic tumor types with much reduced toxicity compared to leptomycin B [64].
In recent years, synthetic inhibitors of XPO1/CRM1, including PKF050-638, CBS9106, and selective inhibitors of nuclear export (SINE), have been developed. CBS9106 was shown to bind XPO1/CRM1, suppress its nuclear export activities, and induce XPO1/CRM1 protein degradation [65, 66]. Treatment with CBS9106 induces cell cycle arrest and apoptosis in a broad spectrum of cancer cells. Oral administration of CBS9106 significantly suppresses tumor growth and prolongs survival without a significant loss of body weight in a xenograft mouse model of multiple myeloma [65]. PKF050-638 originally identified as an inhibitor of HIV Rev protein also interferes with XPO1/CRM1 function [67]. Its N-azolylacrylate scaffold was later adopted by the SINE compounds, a series of structurally related small molecule inhibitors, including KPT-185, KPT-276, KPT-335, KPT-330 (selinexor), KPT-8602 (eltanexor), and SL-801 (felezonexor) [68, 69]. SINE compounds specifically bind to Cys528 in the cargo-binding groove of XPO1/CRM1. In contrast to leptomycin B, the covalent binding of SINE compounds to XPO1/CRM1 is slowly reversible, potentially explaining SINEs’ relatively low toxicity [63, 68]. The development of SINEs has enabled the translational application of nuclear export inhibitors as novel therapeutic agents. A number of SINE compounds have been tested extensively in preclinical settings, exhibiting efficacy in solid tumors [14, 70,71,72,73,74] and hematopoietic malignancies [75,76,77,78,79]. Among the investigated SINEs, KPT-330, KPT-8602, and SL-80 have been advanced to clinical trials. KPT-330 (selinexor) in particular has been evaluated in the majority of the trials and recently received FDA approval for resistant and relapsed multiple myeloma [80].
Clinical trials of XPO1/CRM1 inhibitors
Solid tumors
Single-agent selinexor has shown limited potential in clinical trials for the treatment of solid tumors. A phase 1 study of 189 patients with advanced solid tumors evaluated the safety and efficacy of selinexor (NCT01607905) [81]. Among the 157 evaluable patients, only one complete and six partial responses were observed (4%). Several phase 2 trials further tested single-agent selinexor for the treatment of specific solid tumors. A phase 2 study of selinexor in 56 patients with advanced de-differentiated liposarcoma did not find a significant difference in progression-free survival (PFS) (NCT02606461) [82]. In another phase 2 study of 14 patients with metastatic castration-resistant prostate cancer, single-agent treatment with selinexor led to > 50% reduction in PSA in only two patients. Of the eight patients with a measurable disease at baseline, two had a partial response and four had stable disease as their best radiographic response (NCT02215161). Notably, selinexor treatment was associated with significant toxicities, which limited future clinical application of selinexor in this patient population [83]. Single-agent selinexor was also tested in metastatic triple-negative breast cancer in a phase 2 trial (NCT02402764) with ten heavily pretreated patients. Although fairly well tolerated in these patients, selinexor treatment did not result in objective responses (OR) [84]. In this light, further clinical trials of selinexor and potentially other SINEs in solid tumors should focus on combination therapies. Indeed, several current clinical trials are evaluating combined application of selinexor and other drugs, such as ixazomib (a proteasome inhibitor) in advanced sarcoma (NCT03880123), paclitaxel and carboplatin in advanced ovarian and endometrial cancers (NCT02269293), gemcitabine and nab-paclitaxel in pancreatic cancer (NCT02178436), and several standard chemotherapy regimens for treatment of various advanced solid tumors (NCT02419495).
In contrast to the limited efficacy in solid tumors, selinexor as a single-agent or part of drug combinations has shown promising efficacy in clinical trials of hematopoietic cancers, including acute myeloid leukemia (AML), multiple myeloma, and non-Hodgkin lymphoma (NHL) (summarized in Table 1). Notably, selinexor in combination with several standard therapies has demonstrated superior efficacy in relapsed or refractory multiple myeloma, leading to a recent FDA approval of the selinexor/dexamethasone combination.
Acute myeloid leukemia
Selinexor has been tested in several clinical trials of AML. A phase 1 dose-escalation study examined the safety and efficacy of selinexor in 95 patients with relapsed or refractory (RR) AML (NCT01607892) [85]. Overall, 14% of the patients achieved an OR and 31% showed ≥ 50% decrease in bone marrow blasts. More commonly, however, selinexor has been tested in combination with other cytotoxic or targeted agents. A phase 1 study of selinexor combined with mitoxantrone, etoposide, and cytarabine in 21 patients with RR AML showed an overall response rate (ORR) of 39%, including 4 complete remissions (CR) and 2 complete remissions with incomplete hematologic recovery (CRi) (NCT02299518) [86]. Another phase 1 trial evaluated selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in 20 patients with either newly diagnosed or RR AML (NCT02573363) [87]. An ORR of 70% was observed, with 10 CR, 3 CRi, and 1 PR. In a phase 1b/2 clinical trial of FLT3-mutated refractory AML, selinexor was combined with sorafenib to target XPO1 and FLT3, respectively (NCT02530476). Selinexor/sorafenib combination induced CR or PR in six of 14 patients [88]. Selinexor was also combined with fludarabine and cytarabine for the treatment of pediatric RR acute leukemias and myelodysplastic syndrome (MDS) (NCT02212561). Seven of the 15 evaluable patients achieved CR or CRi, including five with no detectable minimal residual disease [89].
Non-Hodgkin lymphoma
Based on promising preclinical findings, selinexor was also evaluated in clinical trials of NHLs. A phase 1 trial (NCT01607892) tested single-agent selinexor in 79 patients from various NHL subtypes, including DLBCL, Richter’s transformation, mantle cell lymphoma, follicular lymphoma, CLL, and double/triple-hit lymphomas [90]. Twenty-two (31%) of the 70 evaluable patients had an OR, including 4 CR and 18 PR, across all NHL subtypes. These findings suggest that single-agent selinexor has encouraging therapeutic efficacy in patients with RR NHL. A phase 2b study of single-agent selinexor is in progress in 129 patients with RR DLBCL (NCT02227251). In addition to single-agent use, selinexor is currently investigated in clinical trials for the treatment of advanced NHLs, in combination with other agents including RICE (rituximab, ifosfamide, carboplatin, and etoposide) (NCT02471911), R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) (NCT03147885), ibrutinib (NCT02303392), and venetoclax (NCT03955783).
Multiple myeloma
Selinexor has been most extensively tested in multiple myeloma and recently received FDA approval. In an early phase 1 study, selinexor was tested either as a single-agent or in combination with dexamethasone in a cohort of heavily pretreated multiple myeloma patients (NCT01607892) [91]. Single-agent selinexor showed modest efficacy with an ORR of 4%. However, the drug combination was well tolerated with an ORR of 50% (1 CR and 5 PR in 12 patients). Thus, selinexor/dexamethasone combination is active in heavily pretreated multiple myeloma. The combination therapy was further evaluated in a phase 2b trial in a large cohort of heavily pretreated patient (NCT02336815) [92, 93]. These patients had previous exposure to bortezomib, carfilzomib, lenalidomide, pomalidomide, daratumumab, and an alkylating agent and had disease refractory to at least one proteasome inhibitor, one immunomodulatory agent, and daratumumab (triple-class refractory). A partial or better response was observed in 26% of the 122 patients, including two CR. Following the phase 2b trial, selinexor was approved by FDA in combination with dexamethasone, for the treatment of patients with RR multiple myeloma who had received at least four prior therapies and whose disease was refractory to at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody.
A large phase 1/2 trial of selinexor/dexamethasone combination plus other backbone treatments for MM is currently in progress for RR and newly diagnosed patients (NCT02343042). In this trial, the combination of selinexor/dexamethasone/bortezomib has led to an ORR of 63% in a cohort of 42 patients [94]. This preliminary finding has been further extended to a large phase 3 study including 402 RR patients (NCT03110562). Patients in the selinexor/bortezomib/dexamethasone arm had a median PFS of 13.93 months, compared to 9.46 months in patients in the bortezomib plus dexamethasone arm.
Adverse effects in clinical trials
As XPO1 is widely expressed in normal tissues/cells and is required for mitosis, administration of XPO1 inhibitors is expected to cause systemic toxicities. Due to the reversible nature of the XPO1-SINE binding, selinexor is relatively well tolerated in most reported clinical trials. The most common non-hematological adverse events (AEs) are gastrointestinal disturbances, which are primarily grade 1 or 2 but can also be grade 3 [83, 93]. Additionally, asymptomatic hyponatremia and hypokalemia have also been commonly observed. The most common grade 3 or 4 AEs are hematologic, including thrombocytopenia, anemia, and neutropenia. Thrombocytopenia, in particular, is a distinct AE of selinexor due to the impaired thrombopoietin signaling and differentiation of stem cells into megakaryocytes [95]. One SINE compound, KPT-8602, has shown reduced general toxicity in preclinical studies due to limited penetration through the blood-brain barrier [96]. An ongoing clinical trial will determine whether KPT-8602 has equivalent therapeutic efficacy and/or better tolerability in cancer patients (NCT02649790).
Conclusion and future directions
An important aspect of normal cell function, nuclear-cytoplasmic export, is often deregulated in cancers, providing a unique therapeutic opportunity. The recent FDA approval of selinexor for the treatment of RR multiple myeloma lends credence to this therapeutic strategy. Future expansion of the clinical indications of selinexor is contingent upon the success of additional clinical trials. More SINE compounds with improved efficacy and reduced adverse events will be tested and potentially approved for clinical use. In this light, extensive genetic and preclinical studies and well-designed clinical trials are the prerequisite to exploiting the full potential of XPO1 inhibitors in cancer treatment.
Biological functions of XPO1
Identifying the MOA of XPO1 inhibitors is a constantly evolving process. Blockade of nuclear export of tumor suppressor proteins has been postulated as the major MOA. However, recent studies on the diversity and breadth of XPO1 cargoes outline a more complex view. Future endeavors to study the MOA rely on a comprehensive biochemical and genetic characterization of XPO1.
First, XPO1 is frequently overexpressed in human cancers, while XPO1 suppression has been shown to reduce the protein levels of driver oncogenes, such as MYC and EGFR, in multiple cancer types [97,98,99,100]. The detailed mechanisms of reciprocal regulation between XPO1 and driver oncogenes can be dissected using traditional genetic approaches [42]. Second, XPO1 is known to physically interact with hundreds of proteins, many of which may potentially influence its nuclear export activity [101]. Large-scale proteomic experiments coupled with loss-of-function genetic studies can illuminate the roles of the interacting proteins in regulating XPO1 nuclear export activity. Third, the recent deep proteomic study identifying > 1000 proteins exported by XPO1 [15] has significantly broadened our current perspective on XPO1 function. As RNAs have been increasingly recognized as the key XPO1 substrates, high-resolution transcriptomic studies should be carried out in a similar fashion to identify the multiple classes of RNA cargoes. This approach will provide a comprehensive view of XPO1 molecular function. Further comparison of cargoes associated with the wild-type and XPO1 mutant, such as XPO1E571, XPOR749, and XPOD624, will reveal novel gain of function in the mutants, potentially explaining their oncogenic activities, particularly in driving B cell malignancies. These genetic studies of XPO1 will lead to a better understanding of its role in tumorigenesis and the MOA of XPO1 inhibitors.
Mechanisms of drug resistance
With FDA approval of selinexor for the treatment of multiple myeloma, there is an urgent need to investigate the mechanisms of drug resistance. Recurrent genomic mutations detected in patients’ tumors following treatment are associated with resistance. Investigating the causal roles of such mutations in drug resistance requires validation and further examination in cell culture and animal models. Additionally, the mechanisms of resistance can be more broadly studied through CRISPR-based genome-wide library screening [102]. CRISPR screens will identify a comprehensive list of candidate genes causally associated with drug resistance. Potential genetic or epigenetic mechanisms of resistance can be investigated in cell lines and animal models. The clinical relevance of identified candidates will be further examined in human tumor samples.
Synergistic drug combinations
XPO1/CRM1 inhibitors are a unique class of drugs. Thus, their mechanisms of resistance may not be shared by other chemotherapeutic agents. This will provide vast opportunities to combine SINE compounds with other therapeutic modalities. Current clinical development of drug combinations typically relies on empirical testing, which only explores a small fraction of the potential combinations. High-throughput combinatorial screening using drug and/or genetic libraries, on the other hand, can examine millions of combinations in an unbiased manner [103]. As an example, small-molecule combination matrix screens can test XPO1 inhibitors in combination with hundreds of known drugs for systematic identification of synergistic, additive, and antagonistic interactions. Similarly, CRISPR genome-wide library screens can effectively identify potential drug synergies by simultaneous interrogation of thousands of drug targets [104]. Subsequently, therapeutic combinations discovered by such drug/genetic screens may be directly investigated in preclinical animal models and validated in clinical trials. While current preclinical and clinical studies have mainly focused on combining selinexor with existing standard treatments to ensure the timely translation of the findings to clinical applications, future efforts should focus on drug combinations with novel agents, including targeted and immunotherapeutic agents, to improve the efficacy and tolerability.
Biomarkers of therapeutic responses and toxicity
Each human cancer type comprises a highly heterogeneous group of disease. Similar to what has been shown in targeted and immunotherapies, detailed tumor stratification based on genomic, pathohistological, and/or immunological characteristics can help delineate the specific subset of tumors responsive to XPO1 inhibitors. These biomarkers can be identified in preclinical genetic studies and clinical trials and subsequently used in the general patient population to prognosticate the subpopulations most likely to benefit from the therapy. In addition to the tumor-centric approach, pharmacogenomic studies of the patients on their therapeutic response and adverse events will pinpoint major genetic variants associated with inter-individual differences in drug metabolism and response [105, 106]. The availability of biomarkers predictive of therapeutic responses and toxicity will help identify the target patient population and exploit the full potential of XPO1 inhibitors in cancer treatment.
Availability of data and materials
Not applicable
Abbreviations
- AE:
-
Adverse event
- AML:
-
Acute myeloid leukemia
- CR:
-
Complete remission
- CRi:
-
Complete remission with incomplete hematologic recovery
- DLBCL:
-
Diffuse large B cell lymphoma
- MDS:
-
Myelodysplastic syndrome
- MOA:
-
Mechanism of action
- NES:
-
Nuclear export signal
- NHL:
-
Non-Hodgkin lymphoma
- OR:
-
Objective responses
- ORR:
-
Overall response rate
- PFS:
-
Progression-free survival
- RNP:
-
Ribonucleoprotein
- RR:
-
Relapsed or refractory
- SINE:
-
Selective inhibitors of nuclear export
- snRNA:
-
Small nuclear RNA
References
Kimura M, Imamoto N. Biological significance of the importin-beta family-dependent nucleocytoplasmic transport pathways. Traffic. 2014;15(7):727–48.
Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol. 2010;2(10):a000562.
Adachi Y, Yanagida M. Higher order chromosome structure is affected by cold-sensitive mutations in a Schizosaccharomyces pombe gene crm1+ which encodes a 115-kD protein preferentially localized in the nucleus and its periphery. J Cell Biol. 1989;108(4):1195–207.
Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90(6):1041–50.
Hutten S, Kehlenbach RH. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 2007;17(4):193–201.
Becskei A, Mattaj IW. The strategy for coupling the RanGTP gradient to nuclear protein export. Proc Natl Acad Sci U S A. 2003;100(4):1717–22.
Nachury MV, Weis K. The direction of transport through the nuclear pore can be inverted. Proc Natl Acad Sci U S A. 1999;96(17):9622–7.
Fu SC, Huang HC, Horton P, Juan HF. ValidNESs: a database of validated leucine-rich nuclear export signals. Nucleic Acids Res. 2013;41(Database issue):D338–43.
Kim J, McMillan E, Kim HS, Venkateswaran N, Makkar G, Rodriguez-Canales J, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature. 2016;538(7623):114–7.
Gravina GL, Senapedis W, McCauley D, Baloglu E, Shacham S, Festuccia C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol. 2014;7:85.
Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, et al. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc Natl Acad Sci U S A. 1998;95(13):7457–62.
Nakayama R, Zhang YX, Czaplinski JT, Anatone AJ, Sicinska ET, Fletcher JA, et al. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget. 2016;7(13):16581–92.
Tabe Y, Kojima K, Yamamoto S, Sekihara K, Matsushita H, Davis RE, et al. Ribosomal biogenesis and translational flux inhibition by the selective inhibitor of nuclear export (SINE) XPO1 antagonist KPT-185. PLoS One. 2015;10(9):e0137210.
Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol. 2014;74(3):487–95.
Kirli K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, et al. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife. 2015;4.
Thomas F, Kutay U. Biogenesis and nuclear export of ribosomal subunits in higher eukaryotes depend on the CRM1 export pathway. J Cell Sci. 2003;116(Pt 12):2409–19.
Shaw DJ, Eggleton P, Young PJ. Joining the dots: production, processing and targeting of U snRNP to nuclear bodies. Biochim Biophys Acta. 2008;1783(11):2137–44.
Martinez I, Hayes KE, Barr JA, Harold AD, Xie M, Bukhari SIA, et al. An Exportin-1-dependent microRNA biogenesis pathway during human cell quiescence. Proc Natl Acad Sci U S A. 2017;114(25):E4961–70.
Castanotto D, Lingeman R, Riggs AD, Rossi JJ. CRM1 mediates nuclear-cytoplasmic shuttling of mature microRNAs. Proc Natl Acad Sci U S A. 2009;106(51):21655–9.
Bussing I, Yang JS, Lai EC, Grosshans H. The nuclear export receptor XPO-1 supports primary miRNA processing in C. elegans and drosophila. EMBO J. 2010;29(11):1830–9.
Muqbil I, Bao B, Abou-Samra AB, Mohammad RM, Azmi AS. Nuclear export mediated regulation of microRNAs: potential target for drug intervention. Curr Drug Targets. 2013;14(10):1094–100.
Wu J, Bao A, Chatterjee K, Wan Y, Hopper AK. Genome-wide screen uncovers novel pathways for tRNA processing and nuclear-cytoplasmic dynamics. Genes Dev. 2015;29(24):2633–44.
Wickramasinghe VO, Laskey RA. Control of mammalian gene expression by selective mRNA export. Nat Rev Mol Cell Biol. 2015;16(7):431–42.
Okamura M, Inose H, Masuda S. RNA export through the NPC in eukaryotes. Genes (Basel). 2015;6(1):124–49.
Volpon L, Culjkovic-Kraljacic B, Sohn HS, Blanchet-Cohen A, Osborne MJ, Borden KLB. A biochemical framework for eIF4E-dependent mRNA export and nuclear recycling of the export machinery. RNA. 2017;23(6):927–37.
Culjkovic-Kraljacic B, Fernando TM, Marullo R, Calvo-Vidal N, Verma A, Yang S, et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood. 2016;127(7):858–68.
Topisirovic I, Siddiqui N, Lapointe VL, Trost M, Thibault P, Bangeranye C, et al. Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export-competent RNP. EMBO J. 2009;28(8):1087–98.
Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol. 2006;175(3):415–26.
Wozniak R, Burke B, Doye V. Nuclear transport and the mitotic apparatus: an evolving relationship. Cell Mol Life Sci. 2010;67(13):2215–30.
Forbes DJ, Travesa A, Nord MS, Bernis C. Nuclear transport factors: global regulation of mitosis. Curr Opin Cell Biol. 2015;35:78–90.
Arnaoutov A, Azuma Y, Ribbeck K, Joseph J, Boyarchuk Y, Karpova T, et al. Crm1 is a mitotic effector of ran-GTP in somatic cells. Nat Cell Biol. 2005;7(6):626–32.
Torosantucci L, De Luca M, Guarguaglini G, Lavia P, Degrassi F. Localized RanGTP accumulation promotes microtubule nucleation at kinetochores in somatic mammalian cells. Mol Biol Cell. 2008;19(5):1873–82.
Liu Q, Jiang Q, Zhang C. A fraction of Crm1 locates at centrosomes by its CRIME domain and regulates the centrosomal localization of pericentrin. Biochem Biophys Res Commun. 2009;384(3):383–8.
Saulino DM, Younes PS, Bailey JM, Younes M. CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget. 2018;9(30):21289–95.
Chen Y, Camacho SC, Silvers TR, Razak AR, Gabrail NY, Gerecitano JF, et al. Inhibition of the nuclear export receptor XPO1 as a therapeutic target for platinum-resistant ovarian cancer. Clin Cancer Res. 2017;23(6):1552–63.
Shen A, Wang Y, Zhao Y, Zou L, Sun L, Cheng C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009;65(1):153–9 discussion 159-160.
Gupta A, Saltarski JM, White MA, Scaglioni PP, Gerber DE. Therapeutic targeting of nuclear export inhibition in lung cancer. J Thorac Oncol. 2017;12(9):1446–50.
Subhash VV, Yeo MS, Wang L, Tan SH, Wong FY, Thuya WL, et al. Anti-tumor efficacy of Selinexor (KPT-330) in gastric cancer is dependent on nuclear accumulation of p53 tumor suppressor. Sci Rep. 2018;8(1):12248.
Gravina GL, Mancini A, Sanita P, Vitale F, Marampon F, Ventura L, et al. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin [corrected] in prostate cancer models. BMC Cancer. 2015;15:941.
Aladhraei M, Kassem Al-Thobhani A, Poungvarin N, Suwannalert P. Association of XPO1 overexpression with NF-kappaB and Ki67 in colorectal cancer. Asian Pac J Cancer Prev. 2019;20(12):3747–54.
Cosson A, Chapiro E, Bougacha N, Lambert J, Herbi L, Cung HA, et al. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: analysis of the central role of XPO1. Leukemia. 2017;31(7):1625–9.
Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K, et al. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell. 2012;45(2):222–32.
Muralidharan R, Mehta M, Ahmed R, Roy S, Xu L, Aube J, et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci Rep. 2017;7(1):9694.
Jiang JH, Gao Q, Ke AW, Yu Y, Shi GM, Fan J, et al. Prognostic significance of nuclear RNA export factor 3 in hepatocellular carcinoma. Oncol Lett. 2014;7(3):641–6.
Yuen HF, Chan KK, Grills C, Murray JT, Platt-Higgins A, Eldin OS, et al. Ran is a potential therapeutic target for cancer cells with molecular changes associated with activation of the PI3K/Akt/mTORC1 and Ras/MEK/ERK pathways. Clin Cancer Res. 2012;18(2):380–91.
Cui J, Wang L, Ren X, Zhang Y, Zhang H. LRPPRC: a multifunctional protein involved in energy metabolism and human disease. Front Physiol. 2019;10:595.
Yuen HF, Chan KK, Platt-Higgins A, el Dakir H, Matchett KB, Haggag YA, Jithesh PV, Habib T, Faheem A, Dean FA, et al. Ran GTPase promotes cancer progression via met recepto-rmediated downstream signaling. Oncotarget. 2016;7(46):75854–64.
Consortium APG. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818–31.
Taylor J, Sendino M, Gorelick AN, Pastore A, Chang MT, Penson AV, et al. Altered nuclear export signal recognition as a driver of oncogenesis. Cancer Discov. 2019;9(10):1452–67.
Bergsagel PL, Kuehl WM. Comprehensive identification of somatic mutations in chronic lymphocytic leukemia. Cancer Cell. 2011;20(1):5–7.
Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–5.
Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412(6844):341–6.
Hong AL, Tseng YY, Cowley GS, Jonas O, Cheah JH, Kynnap BD, et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat Commun. 2016;7:11987.
Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171(2):481–94 e415.
Tiedemann RE, Zhu YX, Schmidt J, Shi CX, Sereduk C, Yin H, et al. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012;72(3):757–68.
Mathew C, Ghildyal R. CRM1 inhibitors for antiviral therapy. Front Microbiol. 2017;8:1171.
Hamamoto T, Uozumi T, Beppu T. Leptomycins a and B, new antifungal antibiotics. III. Mode of action of leptomycin B on Schizosaccharomyces pombe. J Antibiot (Tokyo). 1985;38(11):1573–80.
Hamamoto T, Gunji S, Tsuji H, Beppu T. Leptomycins a and B, new antifungal antibiotics. I. Taxonomy of the producing strain and their fermentation, purification and characterization. J Antibiot (Tokyo). 1983;36(6):639–45.
Komiyama K, Okada K, Tomisaka S, Umezawa I, Hamamoto T, Beppu T. Antitumor activity of leptomycin B. J Antibiot (Tokyo). 1985;38(3):427–9.
Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer. 1996;74(4):648–9.
Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, Beppu T. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269(9):6320–4.
Koster M, Lykke-Andersen S, Elnakady YA, Gerth K, Washausen P, Hofle G, et al. Ratjadones inhibit nuclear export by blocking CRM1/exportin 1. Exp Cell Res. 2003;286(2):321–31.
Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, et al. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A. 2013;110(4):1303–8.
Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, et al. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69(2):510–7.
Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman JM, et al. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood. 2011;118(14):3922–31.
Saito N, Sakakibara K, Sato T, Friedman JM, Kufe DW, VonHoff DD, et al. CBS9106-induced CRM1 degradation is mediated by cullin ring ligase activity and the neddylation pathway. Mol Cancer Ther. 2014;13(12):3013–23.
Daelemans D, Afonina E, Nilsson J, Werner G, Kjems J, De Clercq E, et al. A synthetic HIV-1 rev inhibitor interfering with the CRM1-mediated nuclear export. Proc Natl Acad Sci U S A. 2002;99(22):14440–5.
Sun Q, Chen X, Zhou Q, Burstein E, Yang S, Jia D. Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal Transduct Target Ther. 2016;1:16010.
Camus V, Miloudi H, Taly A, Sola B, Jardin F. XPO1 in B cell hematological malignancies: from recurrent somatic mutations to targeted therapy. J Hematol Oncol. 2017;10(1):47.
Lee J, Jo DH, Kim JH, Cho CS, Han JE, Kim Y, et al. Development of a patient-derived xenograft model of glioblastoma via intravitreal injection in mice. Exp Mol Med. 2019;51(4):43.
Wang S, Han X, Wang J, Yao J, Shi Y. Antitumor effects of a novel chromosome region maintenance 1 (CRM1) inhibitor on non-small cell lung cancer cells in vitro and in mouse tumor xenografts. PLoS One. 2014;9(3):e89848.
Sun H, Hattori N, Chien W, Sun Q, Sudo M, E-Ling GL, et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer. 2014;111(2):281–91.
Azmi AS, Khan HY, Muqbil I, Aboukameel A, Neggers JE, Daelemans D, et al. Preclinical assessment with clinical validation of selinexor with gemcitabine and nab-paclitaxel for the treatment of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2020;26(6):1338–48.
Green AL, Ramkissoon SH, McCauley D, Jones K, Perry JA, Hsu JH, et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro-Oncology. 2015;17(5):697–707.
Etchin J, Montero J, Berezovskaya A, Le BT, Kentsis A, Christie AL, et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia. 2016;30(1):190–9.
Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122(17):3034–44.
Hing ZA, Mantel R, Beckwith KA, Guinn D, Williams E, Smith LL, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood. 2015;125(20):3128–32.
Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28(1):155–65.
Zhang K, Wang M, Tamayo AT, Shacham S, Kauffman M, Lee J, et al. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol. 2013;41(1):67–78 e64.
XPO1 inhibitor approved for multiple myeloma. Cancer Discov. 2019;9(9):1150–1.
Abdul Razak AR, Mau-Soerensen M, Gabrail NY, Gerecitano JF, Shields AF, Unger TJ, et al. First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol. 2016;34(34):4142–50.
Gounder MM, Somaiah N, Attia S, Chawla SP, Villalobos VM, Chmielowski B, et al. Phase 2 results of selinexor in advanced de-differentiated (DDLS) liposarcoma (SEAL) study: a phase 2/3, randomized, double blind, placebo controlled cross-over study. J Clin Oncol. 2018;36(15_suppl):11512.
Wei XX, Siegel AP, Aggarwal R, Lin AM, Friedlander TW, Fong L, et al. A phase II trial of selinexor, an oral selective inhibitor of nuclear export compound, in abiraterone- and/or enzalutamide-refractory metastatic castration-resistant prostate cancer. Oncologist. 2018;23(6):656–e664.
Shafique M, Ismail-Khan R, Extermann M, Sullivan D, Goodridge D, Boulware D, et al. A phase II trial of selinexor (KPT-330) for metastatic triple-negative breast cancer. Oncologist. 2019;24(7):887–e416.
Garzon R, Savona M, Baz R, Andreeff M, Gabrail N, Gutierrez M, et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood. 2017;129(24):3165–74.
Bhatnagar B, Walker AR, Mims AS, Vasu S, Klisovic RB, Behbehani G, et al. Phase 1 study of selinexor plus mitoxantrone, etoposide, and cytarabine in acute myeloid leukemia. J Clin Oncol. 2018;36(15_suppl):7048.
Wang AY, Weiner H, Green M, Chang H, Fulton N, Larson RA, et al. A phase I study of selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in patients with acute myeloid leukemia. J Hematol Oncol. 2018;11(1):4.
Zhang W, Ly C, Ishizawa J, Mu H, Ruvolo V, Shacham S, et al. Combinatorial targeting of XPO1 and FLT3 exerts synergistic anti-leukemia effects through induction of differentiation and apoptosis in FLT3-mutated acute myeloid leukemias: from concept to clinical trial. Haematologica. 2018;103(10):1642–53.
Alexander TB, Lacayo NJ, Choi JK, Ribeiro RC, Pui CH, Rubnitz JE. Phase I study of selinexor, a selective inhibitor of nuclear export, in combination with fludarabine and cytarabine, in pediatric relapsed or refractory acute leukemia. J Clin Oncol. 2016;34(34):4094–101.
Kuruvilla J, Savona M, Baz R, Mau-Sorensen PM, Gabrail N, Garzon R, et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood. 2017;129(24):3175–83.
Chen C, Siegel D, Gutierrez M, Jacoby M, Hofmeister CC, Gabrail N, et al. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia. Blood. 2018;131(8):855–63.
Vogl DT, Dingli D, Cornell RF, Huff CA, Jagannath S, Bhutani D, et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol. 2018;36(9):859–66.
Chari A, Vogl DT, Gavriatopoulou M, Nooka AK, Yee AJ, Huff CA, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019;381(8):727–38.
Bahlis NJ, Sutherland H, White D, Sebag M, Lentzsch S, Kotb R, et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood. 2018;132(24):2546–54.
Machlus KR, Wu SK, Vijey P, Soussou TS, Liu ZJ, Shacham E, et al. Selinexor-induced thrombocytopenia results from inhibition of thrombopoietin signaling in early megakaryopoiesis. Blood. 2017;130(9):1132–43.
Etchin J, Berezovskaya A, Conway AS, Galinsky IA, Stone RM, Baloglu E, et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia. 2017;31(1):143–50.
Liu Y, Azizian NG, Dou Y, Pham LV, Li Y. Simultaneous targeting of XPO1 and BCL2 as an effective treatment strategy for double-hit lymphoma. J Hematol Oncol. 2019;12(1):119.
Ranganathan P, Kashyap T, Yu X, Meng X, Lai TH, McNeil B, et al. XPO1 inhibition using selinexor synergizes with chemotherapy in acute myeloid leukemia by targeting DNA repair and restoring topoisomerase IIalpha to the nucleus. Clin Cancer Res. 2016;22(24):6142–52.
Gao J, Azmi AS, Aboukameel A, Kauffman M, Shacham S, Abou-Samra AB, et al. Nuclear retention of Fbw7 by specific inhibitors of nuclear export leads to Notch1 degradation in pancreatic cancer. Oncotarget. 2014;5(11):3444–54.
Azmi AS, Li Y, Muqbil I, Aboukameel A, Senapedis W, Baloglu E, et al. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget. 2017;8(47):82144–55.
Martin AP, Jacquemyn M, Lipecka J, Chhuon C, Aushev VN, Meunier B, et al. STK38 kinase acts as XPO1 gatekeeper regulating the nuclear export of autophagy proteins and other cargoes. EMBO Rep. 2019;20(11):e48150.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–7.
Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A. 2014;111(6):2349–54.
Doench JG. Am I ready for CRISPR? A user’s guide to genetic screens. Nat Rev Genet. 2018;19(2):67–80.
Ventola CL. Role of pharmacogenomic biomarkers in predicting and improving drug response: part 1: the clinical significance of pharmacogenetic variants. P T. 2013;38(9):545–60.
Low SK, Takahashi A, Mushiroda T, Kubo M. Genome-wide association study: a useful tool to identify common genetic variants associated with drug toxicity and efficacy in cancer pharmacogenomics. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(10):2541–52.
Acknowledgements
Not applicable
Funding
This work was supported by an NIH Career Transition Award (K22CA207598) to YL.
Author information
Authors and Affiliations
Contributions
NGA and YL wrote the manuscript and contributed equally to the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Azizian, N.G., Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J Hematol Oncol 13, 61 (2020). https://doi.org/10.1186/s13045-020-00903-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00903-4