
Abstract
Background
Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are clinically distinct neurodevelopmental disorders caused by absence of paternally or maternally expressed imprinted genes on chromosome 15q11.2-q13.3 region.
Methods
3331 individuals was recruited from June 2013 to December 2016 under an institutional review board-approved protocol of informed consent. The methylation-specific PCR was employed as a first-tier screening test. The multiplex-fluorescent-labeled STR linkage analysis was carried out to define the underlying genetic mechanisms. The chromosomal microarray analysis was employed to identify chromosomal breakpoints in confirmed cases, and to detect other chromosomal abnormalities in undiagnosed cases. Genetic counseling and recurrence risk assessment were provided to families with affected individuals.
Results
The methylation-specific PCR identified 36 PWS suspected patients and 13 AS suspected patients. UBE3A sequence analysis identified another 1 patient with AS. The STR linkage analysis define the underlying genetic mechanisms. Thirty PWS patients were with paternal deletions on chromosome region 15q11-q13, 5 with isodisomic uniparental disomy and 1 with mixed segmental isodisomic/ heterodisomic uniparental disomy of maternal chromosome 15. Twelve AS patients were with maternal deletions, 1 with isodisomic uniparental disomy and 1 with UBE3A gene mutation. The chromosomal microarray analysis identified chromosomal breakpoints in confirmed cases, and detected chromosomal abnormalities in another 4 patients with clinically overlapped features but tested negative for PWS/AS. Genetic counseling was offered to all families with affected individuals.
Conclusions
Identifying the disorders at early age, establishing the molecular mechanisms, carrying out treatment intervention and close monitoring can significantly improve the prognosis of PWS/AS patients.
Similar content being viewed by others

Background
Prader-Willi syndrome (PWS, OMIM ref. 176,270) and Angelman syndrome (AS, OMIM ref. 105,830) are clinically distinct neurogenetic disorders with multiple phenotypic manifestations. PWS patients present with neonatal hypotonia, poor sucking and weak cry in the postnatal period, delayed psychomotor development and hyperphagia in early childhood, severe obesity, short stature and hypogonadism in adolescents [1]; The initial symptoms of AS overlap with a lot of other disorders, and more characteristic features present later in childhood, including microcephaly, severe developmental delay, gait ataxia and/or tremulousness of the limbs, seizures, absent or severely limited speech, and a unique behavior with happy demeanor [2]. Each syndrome occurs with an estimated prevalence of 1:15,000–1:25,000 live births [3].
The two clinically distinct disorders are both caused by genetic alterations in chromosome region 15q11.2–q13. PWS is attributed to deficiencies of paternally expressed genes, usually as the consequence of paternal deletion (65–70%), maternal uniparental disomy (UPD(15)mat, 20–30%), mutations/ deletions of the imprinting centre (2–5%) and a translocation of the imprinting center (< 1%) [2, 3]. AS arises from molecular defects including maternal deletion (70–75%), paternal uniparental disomy (UPD(15)pat, 2–7%), UBE3A gene mutations (5–10%) and mutations/ deletions of the imprinting centre (3–5%) [3, 4]. Establishing the molecular mechanism will provide information on possible clinical features, prognosis and recurrence risk [5].
Methods
Ethics statement
The study has been approved by Medical Ethics Committee of Guangdong Women and Children Hospital. Written informed consent was obtained from all participants, their parents or legal guardians (in the case of children under 16). Authors had access to information that could identify individual participants, and the information was anonymized prior to submission. All the procedures performed in the study were in accordance with the Declaration of Helsinki.
Patients and samples
Key Laboratory of Inherited Metabolic Diseases under Health Department of Guangdong Province was set up in Guangdong Women and Children Hospital, and has been served as a tertiary referral centre for the province. The centre provides comprehensive genetic counseling, diagnostic and laboratory service for suspected patients and their families. 290 patients suspected for PWS/AS were referred for further diagnosis and genetic confirmation, and 3041 neonates with initial symptoms of PWS/AS were recommended for the genetic screening. The study cohort of 3331 individuals was recruited from June 2013 to December 2016 under an institutional review board-approved protocol of informed consent.
Clinical manifestations
Of the 3331 suspected individuals, 3241 were neonates (≤ 28 days) presented with hypotonia, poor responsiveness, feeding difficulty and weak cry; 49 were infants (1 month to 1 year of age) demonstrated with hypotonia and delayed psychomotor development; another 39 children (1 year to 15 years of age) were referred to the center because of some further characteristic behaviors of PWS or AS. Given that both PWS and AS have clinical overlaps with other diseases, it is difficult to diagnose them solely based on clinical manifestations.
Genetic diagnosis
Blood samples were obtained from all participants and genomic DNA was isolated from the whole blood by standard procedures using the DNA isolation mini kit (ZEESAN, Xiamen, China) and Lab-Aid 820 nucleic acid extraction instrument (ZEESAN, Xiamen, China).
Methylation-specific PCR analysis
Genomic DNA was treated with sulfite using the CpGenome Turbo Bisulfite Modification Kit (Millipore, CA, USA) according to the manufacturer’s instructions. The imprinting gene SNRPN containing a potential imprinting center for a chromosome domain on chr15q11–13. Almost all CpG dinucleotides are methylated on the maternal chromosome and unmethylated on the paternal chromosome. The methylated SNRPN locus was amplified with the MF 5’-TAAATAAGTACGTTTGCGCGGTC-3′ and MR 5’-AACCTTACCCGCTCCATCGCG-3′ to generate the 174 bp methylation product. While the non-methylation primers PF 5′-GTAGGTTGGTGTGTATGTTTAGGT-3′ and PR 5’-ACATCAAACATCTCCAACAACCA-3′, were used to amplify 100 bp of the non-methylated allele.
STR linkage analysis
The multiplex-fluorescent-labeled STR linkage analysis was performed as previously described. [2] The microsatellite markers were selected according to their high heterozygosity and locations in the typical deletion region and adjacent regions. Seven STR markers D15S11, D15S646, D15S817, D15S128, D15S1513, GABRB3, D15S822 located in the typical PWS/AS deletion region of BP1 to BP3, and two loci D15S659, FES in the distal region near telomere. The experimental procedures were as previously described. [2] GeneMapper software (Applied Biosystems) was used for data collection and allele sizing.
Chromosomal microarray analysis
The chromosomal microarray analysis (CMA) was carried out to identify chromosomal breakpoints in confirmed cases. And in undetected cases, it was employed to detect other chromosomal abnormalities. From 2012 to 2014, Agilent’s 8 × 60 K commercial array (Agilent Technologies, CA, USA) was employed for array-based CGH analysis, and Agilent Genomic Workbench Lite Edition 6.5.0.18 software (Agilent Technologies) was used for data analysis. From 2015 onwards, CMA was performed with CytoScan 750 K array (Affymetrix, CA, USA), and Affymetrix Chromosome Analysis Suite software was used for genotype calling, quality control and CNV identification.
Results
Methylation-specific PCR analysis
The PCR products of 174 bp and 100 bp were obtained from methylated and unmethylated alleles of SNRPN gene locus. In 13 patients, the MS-PCR results demonstrated absence of maternal allele at 15q11-q13 or deletion of a methylated CpG island at the SNRPN gene (presented with only 100 bp of paternal fragment), were suspected for AS; 36 patients’ analysis results showed the paternal chromosome is aberrantly methylated at this region (presented with only 174 bp of maternal fragment), and were suspected for PWS (Fig. 1).

MS-PCR analysis of SNRPN gene in PWS/ AS patients. MS-PCR products of 174 bp and 100 bp are amplified from methylated and unmethylated alleles of the SNRPN gene locus respectively. a Lane 1, 1000-bp DNA ladder marker; Lane 2, patient’s father; Lane 3, patient’s mother; Lane 4 and 5, PWS patient from case 2; Lane 6, negative control; Lane 7, PWS positive control; Lane 8, AS positive control; Lane 9, blank control. b Lane 1, 1000-bp DNA ladder marker; Lane 2, patient’s father; Lane 3, patient’s mother; Lane 4 and 5, AS patient from case 38; Lane 6, negative control; Lane 7, PWS positive control; Lane 8, AS positive control; Lane 9, blank control
The MS-PCR results of another 3281 participants presented normal (with both 100 bp and 174 bp fragments). Clinical re-assessments were carried out. For those individuals presented with characteristic features of AS, UBE3A sequence analysis were recommended; For those presented with mild or atypical symptoms, further examinations and adequate follow-up were provided; For those demonstrated with severe manifestations, a careful review of the patients’ histories, clinical features and EEG findings were suggested for differential diagnoses.
STR linkage analysis
The multiplex-fluorescent-labeled STR linkage analysis was carried out to define the underlying genetic mechanism in 49 suspected patients and their parents. 13 patients showed the maternal region deletion at the chromosome 15q11-q13 and were confirmed as AS. Of them, one patient carried two chromosome 15 alleles inherited from one paternal chromosome 15, and was identified as AS with isodisomic uniparental disomy type. Moreover, 36 patients demonstrated with the paternal deletion in this region and were diagnosed as PWS. Of them, 5 patients carried two chromosome 15 alleles inherited from one maternal chromosome 15, and was identified as PWS with isodisomic uniparental disomy type. Another one patient was diagnosed with PWS caused by mixed segmental isodisomic/heterodisomic uniparental disomy of maternal chromosome 15. The results of all the patients tested by STR linkage analysis were listed in Table 1, and some representative cases were shown in Fig. 2.

STR linkage analysis results of PWS (deletion type and UPD type) and AS (deletion type). a The distribution of nine STR locus. Seven STR loci alleles are located in the typical PWS/AS deletion region of BP1 to BP3, and the remaining two are located in the distal region near the telomere. Cen: centromere; Tel: telomere; BP: breakpoint. b Linkage analysis of Case 18 showing the PWS patient has the paternal deletion mutation of chromosome 15 fragment (STR D15S128, D15S646, D15S817, D15S822). c Linkage analysis of Case 19 showing the PWS patient carries mixed segmental isoUPD and heteroUPD of maternal chromosome 15 fragment (STR D15S128, GABRB3, D15S659). d Linkage analysis of Case 40 showing the AS patient has the maternal deletion mutation of chromosome 15 fragment (STR D15S1513, GABRB3, D15S822)
In addition, one AS patient was confirmed by further analysis of UBE3A gene. Table 2 shows the pattern of underlying genetic mechanisms in our cohort.
Chromosomal microarray analysis
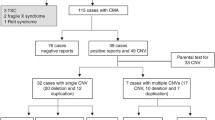
For 21 PWS/AS patients, CMA was carried out to validate previous results and to detect the chromosomal breakpoints. The results were in accordance with previous tests and demonstrated with various deletion fragments. The results of some representative cases were shown in Fig. 3. For another 56 subjects presented with severe clinical manifestations but tested negative for PWS/AS, CMA was employed to detect other chromosomal abnormalities. It identified 14 CNVs in 11 out of 56 (19.6%) patients. Among these CNVs, 5 were classified as pathogenic, 6 as VOUS, and 3 as benign. Two pathogenic CNVs were reported in one patient. Therefore, CMA approach identified another 4 patients in 56 individuals with clinically overlapped features but tested negative for PWS/AS. The causative CNVs and their related syndromes were listed in Table 3. The 1p36 microdeletion syndrome resembles PWS presenting with developmental delays/ intellectual disability, craniofacial dysmorphism, hypotonia and other congenital anomalies [3]. The Mowat–Wilson syndrome is caused by heterozygous mutations in ZEB2 gene on chromosome 2, is associated with severe mental retardation, microcephaly, seizures, short stature, and characteristic facial features that resemble those of AS [3]. The 22q13.3 deletion syndrome may present with nondysmorphic facial features, absent or minimal speech, and moderate to severe developmental delay, sometimes with behavioral features in the autism spectrum, and may present with an AS-like phenotype [3]. Another patient with an AS-like condition carried 17p13.2-pter deletion and 22q13.33-qter duplication, presented with brain abnormalities, developmental delay, facial dysmorphisms, hypotonia and seizures.

CMA results of some representative cases with PWS/AS. a Chromosomal microarray analysis of Case 16 showing the patient has a deletion mutation of chromosome 15. b Chromosomal microarray analysis of Case 19 showing the patient has segmental iso/heteroUPD on chromosome 15
Discussion
The strategies for the analysis of PWS/AS can be affected by many factors, including the arrangement of laboratory services, the coverage of medical insurance and the patterns of referral. [6] In the present study, the MS-PCR approach was employed as a first-tier screening test to detect abnormal parent-specific methylation within the PWS and AS critical region. It is a rapid and convenient platform, which can accurately identify more than 99% of PWS and approximately 80% of AS [2, 6, 7]. UBE3A sequence analysis detects mutations in approximately a further 10% of patients with AS [6]. The multiplex-fluorescent-labeled STRs assay based linkage analysis was carried out to define the underlying genetic mechanisms. It can differentiate the molecular defects between typical deletion and UPD, exactly uniparental heterodisomy [2]. Establishing the mechanism will provide information on the possible clinical features, the prognosis and the recurrence risk [8]. Moreover, CMA was carried out to validate previous results and to detect the chromosomal breakpoints. It is an efficient and sensitive method for precisely detecting CNVs, and SNP-based CMA can directly identify isodisomic UPDs or identical by descents (IBDs) and associated mosaicisms. [9] The strategies adopted in the study for the analysis of PWS/AS demonstrated with satisfying performance. Yet the routine approaches did not include methylation-specific multiplex ligation dependent probe amplification (MS-MLPA) analysis, which can simultaneously detect copy number changes and DNA methylation defects within chromosome 15q11–13 region in a semi-quantitative manner [10], mainly out of the financial consideration. Since the costs of genetic testing for PWS/AS were not covered by the Medicare scheme in China, more cost-effective approaches were employed for the routine detection.
As to the study cohort, the majority of suspected individuals (3241 out of 3331) were neonates (≤28 days) presented with initial symptoms of PWS/AS, such as poor reflexes, sucking and swallowing difficulties. Given that the characteristic signs evolve with age and the initial symptoms overlap with other disorders, it could be difficult for the clinical diagnosis of PWS/AS in early infancy. As previous studies indicated [2, 5, 10, 11], the median age of diagnosis in PWS patients varied between 0.8 and 19.8 years, and in AS patients it was 0.9 to 6.2 years. In the present study, genetic screening was carried out in suspected individuals from neonatal intensive care units. The median age of diagnosis was 12 days in PWS patients and 22 days in AS patients. The prognosis of confirmed patients has improved significantly by early and continued therapies. Furthermore, by carefully analyzing the clinical manifestations of PWS/AS neonates in the present study and retrospectively viewing the special features of Asian patients in previous researches [2, 7, 10,11,12,13], a better understanding of clinical manifestations in Asian PWS/AS patients was learned. It would facilitate the differential diagnosis and early referral, and could result in early diagnosis and better management for PWS/AS patients.
Moreover, in this cohort, 83.3% of PWS were caused by paternal microdeletion, 16.7% by UPD(15)mat; while 85.7% of AS were caused by maternal microdeletion, 7.1% by UPD(15)pat, and 7.1% by UBE3A gene mutation. The distributin pattern of underlying meachanisms in this cohort was compared with those in other published cohorts. In western populations like United States and Germany, about 70–75% of PWS were caused by paternal microdeletion and 20–25% by UPD(15)mat; whereas in Asian regions like Japan, Korea and Taiwan of China, 80% of PWS were caused by paternal microdeletion and 15–20% by UPD(15)mat. The distributin of underlying meachanisms in our cohort of PWS patients was similar to the patterns in Asian PWS cohorts. Besides, in previous published cohorts, 70–75% of AS were caused by maternal microdeletion, 2–7% by UPD(15)pat, 3–5% by IC defect and about 5–10% by UBE3A mutation. The distributin of underlying meachanisms in our cohort of AS patients was similar to the patterns in published AS cohorts, only the proportion of AS caused by maternal microdeletion was comparetively higher than in other cohorts. Microdeletion type was the most common reported mechanism for AS. One of the explanation was chromosome 15q11–13 region harbored multiple low copy repeats (LCRs) that mediated recurrent homologous rearrangement like deletion, duplication and inversion which made this imprinted region as one of the most variable regions in the human genome. Another explanation for its higher proportion might due to ethnic backgorund. The cohort in our study was composed of Chinese Han population, and most of Southern China ancestries. Yet the difference might still be caused by bias in the distributin due to the limited size of the AS patient cohort. Genetic counseling was offered to families with affected individuals, concerning the nature of the diseases, genetic etiology of different molecular classes, interventions and prognosis of PWS/AS patients. Assessment of recurrence risk was carried out based on the genetic mechanism of the proband [4, 5].
Conclusions
A practical set of molecular genetic testing has been adopted for the diagnosis of PWS/AS in the clinical practice of Guangdong Province, and demonstrated with comparatively satisfying performance. A better understanding of clinical manifestations and underlying meachanisms in Chinese PWS/AS patients was learned through this comparetively large cohort of 36 PWS patients and 14 AS patients. Identifying the disorders at early age, establishing the molecular mechanisms, carrying out treatment intervention and close monitoring can significantly improve the prognosis of PWS/AS patients.
Abbreviations
- AS:
-
Angelman syndrome
- CMA:
-
Chromosomal microarray analysis
- IBDs:
-
Identical by descents
- LCRs:
-
Low copy repeats
- MS-PCR:
-
Methylation-specific PCR
- PWS:
-
Prader-Willi syndrome
- STRs:
-
Short tandem repeats
- UPD:
-
Uniparental disomy
- VOUS:
-
Variants of unknown significance
References
Varela MC, Simoes-Sato AY, Kim CA, Bertola DR, De Castro CI, Koiffmann CP. A new case of interstitial 6q16.2 deletion in a patient with Prader-Willi-like phenotype and investigation of SIM1 gene deletion in 87 patients with syndromic obesity. Eur J Med Genet. 2006;49(4):298–305.
Zhang K, Liu S, Feng B, Yang Y, Zhang H, Dong R, Liu Y, Gai Z. Clinical application of an innovative multiplex-fluorescent-labeled STRs assay for Prader-Willi syndrome and Angelman syndrome. PLoS One. 2016;11(2):e0147824.
Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(3):365–76.
Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385–95.
Faundes V, María LS, Aliaga S, Curotto B, Pugin A, Alliende MA. Molecular classes in 209 patients with Prader-Willi or Angelman syndromes: lessons for genetic counseling. Am J Med Genet A. 2015;167A(1):261–3.
Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet. 2010;11:70.
Teng YN, Tsai WH, Wu CJ, Lin SJ, Chen YJ, Kuo PL. Referral diagnosis of Prader-Willi syndrome and Angelman syndrome based on methylation-specific polymerase chain reaction. J Formos Med Assoc. 2002;101:488–94.
Smith A, Hung D. The dilemma of diagnostic testing for Prader-Willi syndrome. Transl Pediatr. 2017;6(1):46–56.
Liu W, Zhang R, Wei J, Zhang H, Yu G, Li Z, Chen M, Sun X. Rapid diagnosis of imprinting disorders involving copy number variation and uniparental Disomy using genome-wide SNP microarrays. Cytogenet Genome Res. 2015;146(1):9–18.
Luk HM, Lo IF. Angelman syndrome in Hong Kong Chinese: a 20 years' experience. Eur J Med Genet. 2016;59(6–7):315–9.
Lo IF, Luk HM, Tong TM, Lai KK, Chan DH, Lam AC, Chan DK, Hau EW, Fung CO, Lam ST. Prader--Willi syndrome: 16-year experience in Hong Kong. J Genet Genomics. 2012;39(4):191–4.
Zhu J, Cao Q, Zhang N, Zhao L. Prader-willi syndrome: a case report and a Chinese literature review. Intractable Rare Dis Res. 2013;2(4):123–6.
Wang P, Zhou W, Yuan W, Huang L, Zhao N, Chen X. Prader-Willi syndrome in neonates: twenty cases and review of the literature in southern China. BMC Pediatr. 2016;16:124.
Acknowledgements
Not applicable
Funding
This study was supported by the Medical Science and Technology Research Project of Guangdong Province (Grant No. A2016225). The funding did not play role in the design of the study, collection, analysis, interpretation of data, or in writing the manuscript.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due individual privacy but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
AY and CL conceived and designed the study. CL, XZ, JW, YZ, AW, JL, SL, JW, LD, JY, HD, LL and XZ performed the genetic diagnosis. CL, XZ, JW and YH analyzed and interpreted data. CL drafted the manuscript and AY revised it. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study has been approved by the Institutional Review Board/ Medical Ethics Committee of Guangdong Women and Children Hospital (IRB reference number: 201701085). Written informed consent was obtained from all participants, their parents or legal guardians (in the case of children under 16).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, C., Zhang, X., Wang, J. et al. Genetic testing for Prader-Willi syndrome and Angelman syndrome in the clinical practice of Guangdong Province, China. Mol Cytogenet 12, 7 (2019). https://doi.org/10.1186/s13039-019-0420-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-019-0420-x