
Abstract
Background
Chromosome 6q duplication syndrome is a chromosome abnormality associated with characteristic phenotypic features such as intellectual disability (ID), short stature, feeding difficulties, microcephaly, dysmorphic features (prominent forehead, downslanting palpebral fissures, flat nasal bridge, tented upper lip, micrognathia, short webbed neck) and joint contractures. Only a few cases of pure partial 6q trisomy have been published and the severity of the phenotype seems to depend on the breakpoint position. Unfortunately, most of these cases were identified using karyotyping or FISH, so breakpoints at the molecular level and thus gene content are not known.
Cases presentation
We report the first two families with an interstitial 6q duplication identified by karyotyping where the gene content and breakpoints were characterized with microarray. In family 1, the 6q22.1q23.2 duplication was detected in a female patient with ID. In family 2, the 6q21q22.33 duplication was identified in a male fetus with multiple congenital malformations. In both families, the duplication seems to show phenotypic heterogeneity and in family 1 also incomplete penetrance suggesting the co-existence of an “additional hit” in affected patients. This “additional hit” was identified in the first family to be a microduplication in 16p11.2, a known susceptibility locus (SL) for neurodevelopmental disorders, that co-segregated with an abnormal phenotype in the affected family members.
Conclusions
Our study shows that interstitial 6q21q23 duplication may represent a private variant that is benign, but may also contribute to developmental disorders of variable expressivity in a “multi-hit” model. Finding the “additional hit” within the family is therefore very important for genetic counseling and assessment of the CNV penetrance within the particular family.
Similar content being viewed by others
Background
The chromosome 6q duplication syndrome is a known chromosome abnormality associated with characteristic phenotypic features such as severe intellectual disability (ID), short stature, feeding difficulties, microcephaly, dysmorphic features (prominent forehead, downslanting palpebral fissures, flat nasal bridge, tented upper lip, micrognathia, short webbed neck) and joint contractures [1–5]. Most reported patients carry unbalanced translocations leading to a partial trisomy 6q and a partial monosomy of another chromosome. In these cases the monosomic region of another chromosome is also of great influence on the patient’s phenotype. There are only several cases of pure partial 6q trisomy and the severity of the phenotype seems to depend on the breakpoint position. In general, terminal duplications with co-existing monosomy of another chromosome region cause severe phenotypes whereas patients with pure interstitial duplications seem to be only mildly affected [6–8]. Unfortunately the breakpoints of the majority of published cases were assessed with karyotyping or FISH, which hampers finding critical regions for ID and other features [9] so the genotype-phenotype studies in cases characterized by microarray are of great value [10].
We report on two families with an interstitial imbalance involving region 6q21q23: a familial duplication of 6q22.1q23.2 and one of 6q21q22.33. This is the first report where the gene content and breakpoints of a microscopically visible interstitial 6q duplication was characterized with microarray. In these families, some individuals carrying the imbalance did not show any structural anomalies or ID associated with trisomy 6q as described before, suggesting incomplete penetrance.
Case presentation
Family 1
A 23 year-old pregnant woman was referred to the department of Clinical Genetics due to isolated ID and a familial interstitial duplication on chromosome 6 characterized by karyotyping in the past. This duplication was discovered in 1983 in a family member that was known to suffer from ID, developmental delay (DD) and seizures. The observed ID in this family varied from very mild (low degrees and learning problems) to more severe (accompanied by DD and seizures).
The partner of the pregnant patient was known to have ID as well. He was an adopted child. His biological brother was said to have ID as well. Neither further family history, nor parental samples were available. Because of the pregnancy, our patient and her partner were investigated with genomic SNP array (Illumina HumanCytoSNP-12) to study their chromosomal constitution.
The pregnant patient carried an interstitial ~16.1 Mb 6q duplication and the array testing additionally revealed a susceptibility locus (SL) for neurodevelopmental disorders: a ~580 kb 16p11.2 microduplication (arr[hg19] 6q22.1q23.2(116,411,005-132,541,408)x3,16p11.2(29,595,483-30,174,926)x3). The partner of the pregnant patient was shown to carry another SL for neurodevelopmental disorders, a ~806 kb 16p11.2 microdeletion (arr[hg19] 16p11.2(28,475,873-29,282,147)x1).
Because both partners had abnormal array results, prenatal diagnosis was advised. Amniocentesis was performed at 15 5/7 weeks of gestation and diagnostic array testing on uncultured amniotic fluid cells showed only the interstitial 6q duplication (Fig. 1). In order to enable clinical interpretation of this result, 4 other family members of the pregnant woman, two healthy and two affected (one mild and the other severe (the one that was karyotyped in 1983, see above)), were tested with SNP array. Two healthy relatives turned out to carry only the 6q duplication, whereas the two affected relatives carried both the 6q duplication and the 16p11.2 microduplication.

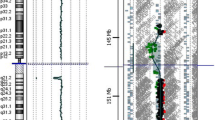
Family 1: Log Ratio plots of Illumina CytoSNP-12: a – partial chromosome 6q plot showing the 6q duplication in family 1: the mother and the fetus carry a 6q22q23 duplication. The arrows indicate the duplicated region. b - partial chromosome 16p plot showing the imbalances in 16p11: the father carries 16p11 microdeletion, the mother 16p11 microduplication, whereas the fetal array profile is normal for both 16p regions. The arrows indicate the imbalances in parental samples
As the fetus inherited only the 6q duplication, in the absence of SL, the prognosis of the fetal phenotype may be favorable. The second trimester fetal ultrasound investigations showed no structural anomalies. A healthy baby boy without any dysmorphic features was born and there were no developmental abnormalities noted so far (follow up at the age of 1 year).
Family 2
A 34 year-old pregnant woman was referred to a clinical geneticist after suspicion of fetal esophageal atresia at third trimester ultrasound investigations, based on polyhydramnios and lack of visualization of the stomach. This was the first pregnancy of non-consanguineous parents. Fetal karyotype (amniotic fluid) revealed an interstitial duplication on chromosome 6. SNP-array (Affymetrix CytoScan HD) was performed, showing a 15.3 Mb duplication (arr[hg19]6q21q22.33(113,465,762-128,774,807)x3) (Fig. 2). Given the combination of the duplication along with fetal structural abnormalities, the parents opted for termination of pregnancy at 33 weeks gestational age. The male fetus had normal biometry: weight 2593 g, height 49 cm, occipitofrontal circumference (OFC) 29 cm. Dysmorphic facial features were noted: short nose with anteverted nares and long eyelashes. Fetal autopsy confirmed the esophageal atresia and revealed additional structural abnormalities: an abnormal pulmonary lobulation (2 lobes on right lung and 1 lobe on left lung), a Meckel’s diverticulum, bilateral hydronephrosis, pancreatic ectopia, and neuronal heterotopia of the cerebellum located on the dentate nuclei.

Family 2: a -Karyotype in standard resolution for chromosome 6, showing the interstitial duplication (arrow). b -array results (Oligo aCGH Agilent 60 K) for chromosome 6 in family 2 showing duplication of 6q present in family 2. c- The duplicated region and its gene content
Familial studies revealed that the mother was carrier of the same interstitial duplication on chromosome 6 (karyotyping and Oligo aCGH Agilent 60 K), while her parents had normal karyotypes. No additional copy number variation was found in both the mother and the fetus. The mother had previously experienced learning difficulties, dyslexia and was known with a scoliosis. Cardiac ultrasound showed no abnormalities. No cerebral or abdominal imaging was performed. She works as a nurse assistant in a hospital.
Discussion
In many laboratories over the world, genomic array testing is now a first-tier cytogenetic test. The interpretation of array results is mainly based on comparisons to CNVs in control and patient populations [11]. Comparison to previously reported cases with microscopically visible chromosome anomalies, however, is problematic. The breakpoints of the majority of published cases were assessed with karyotyping or FISH, which hampers the genotype-phenotype correlation studies as these breakpoints often are not accurate enough to compare them with array results [9]. In the karyotyping era a de novo unbalanced euchromatic chromosome aberration used to be almost always interpreted as pathogenic, however rare inherited variants were assumed to be benign and interpreted as unbalanced euchromatic abnormalities without phenotypic consequences (if inherited from a normal parent) [12–17]. At that time susceptibility CNVs (variants of variable expressivity and phenotypic heterogeneity) [18, 19], which expression seems to depend on other (genetic and/or environmental) factors, were less commonly known.
Phenotypes of 6q21q23 duplication carriers
The literature searched showed only 12 cases of interstitial duplications in chromosome region 6q21-6q23 (Table 1, Fig. 3) [7, 20–26]. The phenotype show variability and as shown in Fig. 3 not all duplication cases overlap with the families presented here. Congenital malformations seem to be infrequent, with only few patients reported to have variable cardiac anomalies (right ventricular hypertrophy, cardiomegaly, ventricular septal defect, pulmonary stenosis) [24, 25], and one suspicion of a corpus callosum agenesis [24]. The 6q duplications in the current families seem to overlap with the ISCA case and patients published by Goh et al., Pratt et al. and Zneimer et al. [20, 24, 25], although the breakpoints based on karyotyping could be highly imprecise. All these patients presented with DD/mild ID and have common craniofacial features with constant hypertelorism and frequent mid-face hypoplasia with anteverted nares [20, 24, 25]. Further, the two cases reported by Pazooki et al. that only partially overlap with the duplication in family 2 also presented DD and similar dysmorphic features (hypertelorism and anteverted nares), but no congenital malformation.

Breakpoints comparison (in GRCh37/hg19) in cases with dup 6q21q23 where molecular testing was done (minimal breakpoints based on FISH/YAC or DNA markers or given by the authors of original papers, if appropriate lifted from other genome builds)
Possible candidate genes for DD/ID in 6q21q23
Although the duplicated regions are large, we conducted a gene content study to search for candidate genes that may explain the ID/learning problems in presented families. Gene lists were generated using Nexus BioDiscovery Copy Number 7.5 and the data were uploaded and analyzed with QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.ingenuity.com). A list of genes and proteins related to ID was generated using the Knowledge Base of IPA. In case of the 6q duplication in family 1, we did not find any candidate gene that may explain the ID when duplicated, which may support our hypothesis that the 6q duplication seems to be benign if present alone. However, the proximal region that was duplicated in family 2, but not in family 1 seems to have an interesting gene content. It contains four OMIM genes: MARCKS, HDAC2, HS3ST5 and FRK, all involved in brain function and development [27–30]. Interestingly it was shown that MARCKS expression is significantly elevated (45 %) in the hippocampus of mice, which exhibit impaired hippocampus-dependent learning [27] and neuron-specific over-expression of HDAC2 decreased dendritic spine density, synapse number, synaptic plasticity and memory formation [31]. Since all four genes are involved in neural development, one could speculate that if their expression is dosage dependent, their duplication may play a role in neurological phenotypes like ID, DD and brain malformations. To our knowledge, so far, there are no cases of whole gene duplications in MARCKS, HDAC2 and HS3ST5 in control populations (neither in Decipher Population CNVs, Toronto Database of Genomic Variants, nor our in house controls), and there is only one family published by Pazooki et al. carrying a smaller duplication, but overlapping also with MARCKS, HDAC2, HS3ST5 (Fig. 3) [26]. Unfortunately these data are not enough evidence to claim that duplication of MARCKS, HDAC2, HS3ST5 causes learning problems or ID/DD. The duplication of these genes remains a variation of unknown clinical significance, however it cannot be excluded that it may contribute to learning problems when also other genes involved in neurological phenotypes are deregulated.
The second hit hypothesis
The variability in phenotypes in patients shown in Table 1 could be explained by different chromosome breakpoints, however the duplications in the current families show variability within a family as well. This cannot be explained by different gene content, but it may be a consequence of the presence of additional anomalies in affected individuals.
The two-hit model has been introduced to explain the phenotypic variability in genetic disorders [18, 19]. The presence of the additional hit may modify phenotype, either acting in an additive manner (independently adding new features), or by participation in the same or similar biochemical pathway causing more severe phenotypes [32]. And indeed in family 1, the 6q22.1q23.2 duplication seems to be an euchromatic variant without phenotypic consequences, as only the individuals carrying the 6q duplication and the additional susceptibility CNV showed abnormal phenotypes. Family 1 shows very high penetrance of the susceptibility locus, when both chromosome aberrations are present. Although this observation can be reassuring for the fetus, we found prediction of the fetal prognosis still problematic since the father of the fetus carried a susceptibility locus for neurodevelopmental disorder, but the paternal “additional hit” remained unknown. The fetus did not inherit the paternal SL, nevertheless the paternal “additional hit” may have been inherited and may influence the intellectual development of the child.
In family 2, the interstitial 6q duplication seems to show highly variable expression in the mother and fetus. The fetus had dysmorphic features similar to reported cases in the literature and multiple congenital malformations that were not described before. The breakpoints and gene content were identical in the mother, who only had learning difficulties in childhood. There are no healthy carriers of this duplication in this family and the de novo occurrence of this chromosomal duplication in the mother of the fetus may be an argument for its causality for the phenotype. The “second hit” has not been identified by microarray in this family and remains unresolved. Genetic counseling for a future pregnancy remains problematic: it is doubtful whether it is reasonable to offer an invasive prenatal test, if the “additional hit” is still unknown and if no fetal ultrasound abnormalities are present. Given these limitations, the couple has opted for preimplantation diagnosis.
Conclusions
Finding a rare euchromatic variant or a susceptibility locus is challenging in both postnatal and prenatal settings. Our study shows that the 6q22.1q23.2 duplication may represent a private variant that is benign when present alone, but that may act as the “additional/second hit” in SL carriers. We suggest that also large benign CNVs can serve as “an additional hit” and therefore our study supports the opinion of Thomas Liehr that benign variants cannot be fully neglected in genetic analysis [17]. The presented family 1 supports “the multi-hit hypothesis” showing that the penetrance of such a SL in families known to carry an “additional hit” can be much higher than predicted [33], which may explain the conflicting conclusions on the association with an abnormal phenotype in the papers on SL [34–36]. Finding the “additional hit” within the family is therefore very important for genetic counseling and the assessment of the SL penetrance within the particular family.
Abbreviations
CNV, copy number variation; DD, developmental delay; FISH, fluorescent in situ hybridization; ID, intellectual disability; OFC, occipitofrontal circumference; SL, susceptibility locus
References
Dallapiccola B, Bricarelli FD, Quartino AR, Mazzilli MC, Chisci R, Gandini E. Delineation of syndromes due to partial 6q imbalances. Trisomy 6q21 leads to qter and monosomy 6q221 leads to qter in two unrelated patients. Acta Genet Med Gemellol (Roma). 1978;27:57–66.
Tipton RE, Berns JS, Johnson WE, Wilroy Jr RS, Summitt RL. Duplication 6q syndrome. Am J Med Genet. 1979;3(4):325–30.
Turleau C, de Grouchy J. Trisomy 6qter. Clin Genet. 1981;19(3):202–6.
Pivnick EK, Qumsiyeh MB, Tharapel AT, Summitt JB, Wilroy RS. Partial duplication of the long arm of chromosome 6: a clinically recognisable syndrome. J Med Genet. 1990;27(8):523–6.
Uhrich S, FitzSimmons J, Easterling TR, Mack L, Disteche CM. Duplication (6q) syndrome diagnosed in utero. Am J Med Genet. 1991;41(3):282–3.
Cappon SL, Duncan AM, Khalifa MM. Interstitial 6q duplication in an adult male without growth delay or severe mental retardation. Med Sci Monit. 2000;6(3):581–5.
Henegariu O, Heerema NA, Vance GH. Mild “duplication 6q syndrome”: a case with partial trisomy (6)(q23.3q25.3). Am J Med Genet. 1997;68(4):450–4.
Zweier C, Trautmann U, Ekici A, Rauch A. A 15 Mb duplication of 6q24.1-q25.3 associated with typical but milder features of the duplication 6q syndrome. Eur J Med Genet. 2008;51(4):358–61.
Tabet AC, Aboura A, Gerard M, Pilorge M, Dupont C, Gadisseux JF, et al. Molecular characterization of a de novo 6q24.2q25.3 duplication interrupting UTRN in a patient with arthrogryposis. Am J Med Genet A. 2010;152A(7):1781–8.
Sheth F, Trivedi S, Andrieux J, Blouin JL, Sheth J. Pure interstitial dup(6)(q22.31q22.31) - a case report. Ital J Pediatr. 2015;41:5.
Carreira IM, Ferreira SI, Matoso E, Pires LM, Ferrao J, Jardim A, et al. Copy number variants prioritization after array-CGH analysis - a cohort of 1000 patients. Mol Cytogenet. 2015;8:103.
Kowalczyk M, Srebniak M, Tomaszewska A. Chromosome abnormalities without phenotypic consequences. J Appl Genet. 2007;48(2):157–66.
Barber JC. Directly transmitted unbalanced chromosome abnormalities and euchromatic variants. J Med Genet. 2005;42(8):609–29.
Barber JC, Joyce CA, Collinson MN, Nicholson JC, Willatt LR, Dyson HM, et al. Duplication of 8p23.1: a cytogenetic anomaly with no established clinical significance. J Med Genet. 1998;35(6):491–6.
Barber JC, Mahl H, Portch J, Crawfurd MD. Interstitial deletions without phenotypic effect: prenatal diagnosis of a new family and brief review. Prenat Diagn. 1991;11(6):411–6.
Liehr T. Benign & Pathological Chromosomal Imbalances, 1st Edition Microscopic and Submicroscopic Copy Number Variations (CNVs) in Genetics and Counseling. Academic Press; 2013.
Liehr T. Cytogenetically visible copy number variations (CG-CNVs) in banding and molecular cytogenetics of human; about heteromorphisms and euchromatic variants. Mol Cytogenet. 2016;9:5.
Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42(3):203–9.
Veltman JA, Brunner HG. Understanding variable expressivity in microdeletion syndromes. Nat Genet. 2010;42(3):192–3.
Goh DL, Tan AS, Chen JY, Van Den Berghe JA. Dysmorphic sibs trisomic for the region 6q22.1-- > 6q23.3. J Med Genet. 2000;37(11):889–92.
Arthur EI, Zlotogora J, Lerer I, Dagan J, Marks K, Abeliovich D. Transient neonatal diabetes mellitus in a child with invdup(6)(q22q23) of paternal origin. Eur J Hum Genet. 1997;5(6):417–9.
Causio F, Fischetto R, Carnevale F, Pansini A, Rocchi M. Trisomy 6q syndrome: a case with a < < de novo > > 6q23 tandem duplication. Genet Couns. 2001;12(2):145–50.
Temple IK, Gardner RJ, Robinson DO, Kibirige MS, Ferguson AW, Baum JD, et al. Further evidence for an imprinted gene for neonatal diabetes localised to chromosome 6q22-q23. Hum Mol Genet. 1996;5(8):1117–21.
Pratt VM, Roberson JR, Weiss L, Van Dyke DL. Duplication 6q21q23 in two unrelated patients. Am J Med Genet. 1998;80(2):112–4.
Zneimer SM, Ziel B, Bachman R. Partial trisomy of chromosome 6q: an interstitial duplication of the long arm. Am J Med Genet. 1998;80(2):133–5.
Pazooki M, Lebbar A, Roubergues A, Baverel F, Letessier D, Dupont JM. Pure familial 6q21q22.1 duplication in two generations. Eur J Med Genet. 2007;50(1):60–5.
McNamara RK, Hussain RJ, Simon EJ, Stumpo DJ, Blackshear PJ, Abel T, et al. Effect of myristoylated alanine-rich C kinase substrate (MARCKS) overexpression on hippocampus-dependent learning and hippocampal synaptic plasticity in MARCKS transgenic mice. Hippocampus. 2005;15(5):675–83.
Jawerka M, Colak D, Dimou L, Spiller C, Lagger S, Montgomery RL, et al. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 2010;6(2):93–107.
Mochizuki H, Yoshida K, Gotoh M, Sugioka S, Kikuchi N, Kwon YD, et al. Characterization of a heparan sulfate 3-O-sulfotransferase-5, an enzyme synthesizing a tetrasulfated disaccharide. J Biol Chem. 2003;278(29):26780–7.
Chandrasekharan S, Qiu TH, Alkharouf N, Brantley K, Mitchell JB, Liu ET. Characterization of mice deficient in the Src family nonreceptor tyrosine kinase Frk/rak. Mol Cell Biol. 2002;22(14):5235–47.
Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60.
Kumar RA. Two-hit wonder: a novel genetic model to explain variable expressivity in severe pediatric phenotypes. Clin Genet. 2010;78(6):517–9.
Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. 2013;15(6):478–81.
Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505(7483):361–6.
Hashemi B, Bassett A, Chitayat D, Chong K, Feldman M, Flanagan J, et al. Deletion of 15q11.2(BP1-BP2) region: Further evidence for lack of phenotypic specificity in a pediatric population. Am J Med Genet A. 2015;167A(9):2098–102.
Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, Boute O, et al. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet. 2015;58(3):140–7.
Acknowledgements
We kindly thank the families for their participation in this study.
Funding
There was no funding available for this study.
Availability of data and materials
CNVs found in family 1 and 2 are included within the article itself. Additionally the dataset including CNVs of family 2 is available in the DECIPHER database https://decipher.sanger.ac.uk/ (Patient 303543). Family 1 did not give a permission to include their data in databases.
Authors’ contributions
MIS conceived the study, made substantial contributions to conception and design, analysis and interpretation of genomic data. LvZ made substantial contributions to acquisition of data, analysis and interpretation of genomic data and participated in the drafting of the manuscript. FP participated in the drafting of the manuscript and coordinated the genetic counseling of the patients. SB and IJPvH made substantial contributions to acquisition of data and carried out analysis and interpretation of data. MK and JC performed pre-test counseling, invasive testing, perinatal management and contributed to the acquisition of phenotypic data (prenatal ultrasound scans). DVO and AK carried literature search and interpretation of data. AK carried out the analysis with IPA. MIS, LvZ, SB, FP and KEMD were responsible for the final diagnoses and reports. KEMD coordinated the genetic counseling of the patients and together with MIS supervised the study. All authors revised the manuscript critically for important intellectual content, approved the final version and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Patients undergoing prenatal diagnosis at Erasmus MC are informed that we may investigate (publish) their medical data as long as all data remained anonymized. The family followed-up at CHU Lille has been informed of this publication. Each patient had the opportunity to object to their inclusion (opting out). No objections were made to this publication. All presented data are anonymized and do not allow identification of the individual patients.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Srebniak, M.I., van Zutven, L.J.C.M., Petit, F. et al. Interstitial 6q21q23 duplication - variant of variable phenotype and incomplete penetrance or benign duplication?. Mol Cytogenet 9, 43 (2016). https://doi.org/10.1186/s13039-016-0253-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-016-0253-9