
Abstract
Background
Interstitial duplications distal to 15q13 are very rare.
Case Presentation
Here, we reported a 14-year-old boy with severe short stature, delayed bone age, hypogonadism, global developmental delay and intellectual disability. His had distinctive facial features including macrocephaly, broad forehead, deep-set and widely spaced eyes, broad nose bridge, shallow philtrum and thick lips. A de novo 6.4 Mb interstitial duplication of 15q15.3q21.2 was detected by chromosomal microarray analysis. We compared our patient’s clinical phenotypes with those of several individuals with overlapping duplications and several candidate genes responsible for the phenotypes were identified as well.
Conclusion
The results suggest a novel contiguous gene duplication syndrome characterized with shared features including short stature, hypogonadism, global developmental delay and other congenital anomalies.
Similar content being viewed by others


Background
Microdeletions and microduplications are recurrent for the proximal region of 15q. Five common breakpoints (BP1–BP5) exist in the 15q11.2-q13 region. Deletions between BP1–BP3 are responsible for the Prader-Willi and Angelman syndromes, depending on the parent-of-origin, whereas the reciprocal duplications of the region, particular the gains of maternal copy of 15q11.2-q13, are known as 15q duplication syndrome (Dup15q) characterized by global developmental delay, autism spectrum disorder and epilepsy [1–3]. 15q13.3 microdeletions and microduplications between BP4–BP5 are enriched in neurodevelopmental and neuropsychiatric disorders [4, 5]. On the other end of the 15q, the terminal trisomy or tetrasomy of 15q, including IGF1R gene, is named as 15q26 overgrowth syndrome consisting of overgrowth, learning difficulties, distinctive facial features and multiple congenital anomalies [6]. The monosomy of 15q26 shows the opposite growth features, such as pre- and postnatal growth retardation, as well as some different dysmorphic features [7]. A few proximal microdeletions at 15q25.2 have been reported to be associated with congenital diaphragmatic hernia, cognitive deficits and Diamond-Blackfan anemia and some more distal deletions to neurodevelopmental and neuropsychiatric disorders [8–11]. Whereas duplications distal to 15q13 are very rare. Elcioglu (1997) described a patient with hypogonadism, skeletal anomalies, Marfan-like features, developmental delay and intellectual disability, who carried a de novo interstitial inverted duplication involving bands of 15q13.3-q21.3 [12]. Herr (1983) reported a patient with a de novo interstitial duplication of 15q14-q21.1, who presented with hypogonadism, skeletal problems, short stature, delayed bone age, global developmental delay and intellectual disability [13]. Both of the duplications were uncovered by high-resolution G-banded cytogenetics analysis, so the precise locations and sizes of the duplications were not determined [12, 13]. Here, we reported a de novo 6.4 Mb interstitial duplication of 15q15.3q21.1 in a 14-year-old boy with hypogonadism, short stature, global developmental delay and multiple congenital anomalies.
Case presentation
The proband is the fourth child of healthy unrelated parents with negative family history. His siblings are all healthy. Intrauterine growth retardation was noticed by ultrasound examination at 7 months of pregnancy. He was born by vaginal delivery at 38 weeks of gestation. Birth weight was 2.9 kg (<-1SD), length 47 cm (<-2SD) and head circumference 34 cm. Apgar scores were all 9. No feeding difficulty was noted at all times.
The development milestones were delayed: he raised his head at 4 months of age, sat at 9 months and independently walked at 1 year 6 months. Language development was significantly delayed. The patient was examined at the age of 14 years. His height was 140 cm (<-2SD), weight 33.5 kg (<-2SD) and head circumference 54 cm, which indicated persistent failure to thrive. He had moderate intellectual disability and showed poor performance in the elementary school. His distinctive facial features were characterized by macrocephaly, coarse face, broad forehead, deep-set and widely spaced eyes, strabismus, broad nose bridge, shallow and short philtrum and thick lips. He had severe short stature and hands X-ray showed delayed bone age (Fig. 1). Yet his growth hormone level (6.19 ng/ml) and level after provocation tests (11.6 ng/ml) were all normal. Pituitary magnetic resonance imaging and thyroid hormone level were also normal. Micropenis, small testes and low testosterone were detected. No additional abnormalities was noticed.

The probandat 14 years of age. Note relative macrocephaly, coarse face, broad forehead, deep-set and widely spaced eyes, strabismus, broad nose bridge, shallow philtrum, thick lips. Hand X-ray showed delayed bone age.
Methods
Chromosomal microarray analysis
Chromosomal microarray analysis was performed for the proband and both parents by Affymetrix Cytoscan HD Array (Affymetrix, USA). Genomic DNA was extracted from peripheral blood using a commercial kit (Qiagen). The labeling and hybridization procedures were performed following manufacturer’s instructions. The raw data of chromosomal microarray was analyzed by Affymetrix Chromosome Analysis Suite Software.
Confirmation of 15q15.3q21.2 duplication
The duplication was further confirmed using quantitative real-time PCR analysis. Primer sequences and descriptions were included in Additional file 1: Table S1.
Results
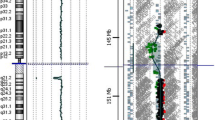
A 6.4Mb duplication at 15q15.3q21.2 (chr15: 44,143,547–50,572,601) was detected by CMA (Fig. 2). Parental tests were normal. Thus, the proband carried a de novo copy number variant. The duplication was further confirmed by quantitative real-time PCR analysis (data not shown).

Affymetrix cytoscan HD array analysis including weighted log2 ratio (upper), copy number state (middle) and allele peaks (lower) are shown for chromosome 15. The result shows duplication at 15q15.3q21.2. The genomic coordinates (hg19): chr15:44,143,547–50,572,601. The copy number gain region is denoted by a blue bar
Discussion
Interstitial microdeletions and duplications involving 15q except for the 15q11-q13 region were very rare. Here, we reported a 14-year-old boy with severe short stature, delayed bone age, hypogonadism, global developmental delay, intellectual disability and distinctive facial features, who carried a de novo 6.4 Mb duplicationon 15q15.3q21.2. No other clinical significant CNVs were detected in this patient. No duplication at this interval was reported in DGV. It was reasonable to suggest that this de novo duplication was likely pathogenic and mostly responsible for the patient’s clinical condition. Next we searched databases for duplications overlapping with this region. We identified 3 cases from DECIPHER, 1 case from ISCA, 2 cases from literatures. The locations and sizes of these duplications are depicted in Fig. 3 and their clinical information detailed in Table 1.

Top panel shows a genome view of all duplication cases (blue colored custom tracks) relative to the genomic coordinates at 15q13.3q22 region, extracted from Human Genome Build 37 (hg19). Red block diagram represents critical region of 15q21.1. Bottom panel shows the zoomed-in 15q21.1 region encompassing candidate genes. Note: Two patients reported by Herr and Elcioglu were detected by high resolution G-banding analysis, therefore the genomic coordinates of the relevant bars were approximate locations.
Elcioglu (1997) [12] and Herr (1983) [13] each reported a case with a de novo interstitial duplication on 15q13.3q21.3 and 15q14q21.1 respectively. The Decipher case 260222 largely overlapped with the duplication detected in our patient. Our patient had the smallest duplication among these four patients with large duplication centered on 15q21.1. We noticed that three patients (our patient, Decipher case 260222 and the patient reported by Herr) had strikingly similar clinical features including hypogonadism, short stature, delayed bone age, global developmental delay and intellectual disability. Interestingly, the patient reported by Elcioglu shared most of similar phenotypes, but presented with Marfan-like features such as tall stature. All duplications were de novo except for Decipher case 260222 without parental tests and not reported in DGV database, as well as these patients had similar clinical conditions especially hypogonadism and skeletal problems, which further suggested a likely pathogenic nature of these duplications.
To further support this notion, we identified several candidate genes responsible for complex clinical phenotypes. The FBN1 (134797) gene is involved in these duplications. As we know that different mutations of FBN1 gene cause different dominant genetic diseases characterized with opposite stature. For example, acromicric dysplasia caused by FBN1 mutations is characterized by severe short stature, short limbs, delayed bone age, stiff joints and facial dysmorphism [14]. Weill-Marchesani syndrome caused by FBN1 mutations consists of short stature, eye abnormalities, unusually brachydactyly, joint stiffness and heart disease [15]. Marfan syndrome caused by FBN1 mutations on the other hand is charaterized by tall and slender stature with arachnodactyly, as well as ocular and cardiovascular defects [16]. Thus, skeletal anomalies could be explained by FBN1 gene duplication at this interval.
Other than the FBN1 gene that was known to cause skeletal problems, no other known disease gene at this interval could explain all clinical phenotypes of these patients. Smaller overlapping duplications shared by Decipher cases 250560 and 303564 and an ISCA case 1602971 involved the NOX family genes such as DUOX2, DUOXA2, DUOXA1 and DUOX1. The loss of function mutations in these genes could cause permanent congenital hypothyroidism [17–20]. However, thyroid examinations of our patient and the one reported by Herr were both normal and other patients were lack of this information, it may be inferred that the clinical consequence of genes duplication was not known. And there was no additional evidence to support the pathogenicity of these small duplications.
Among other genes at the interval, we identified the EID1 gene as an interesting candidate gene. EID1 was shown to be expressed ubiquitously in human tissues with high expression in neuron, cardiac and skeletal muscle [21, 22]. Liu et al. generated an EID1 transgenic mouse model that exhibited neuron-specific overexpression of human EID1 gene in the brain, and overexpression of EID1 reduced hippocampal long-term potentiation and impaired spatial learning and memory function [23]. EID1 tightly interacted with CBP/p300 in the nuclei and had been identified as an inhibitory protein of CBP/P300 [23]. CBP/p300, a family of CREB-binding proteins, was associated with Rubinstein–Taybi syndrome characterized by short stature, intellectual disability, developmental delay and distinctive facial features [24]. Overexpression of EID1 in human brain could negatively regulate CBP/p300 activity and lead to the impairment of neuronal function. In addition, EID-1 was known to be co-expressed with the Steroidogenic factor-1 (SF-1) which played a critical role in adrenal and reproductive development and function [25, 26]. Park et al. provided convincing evidence demonstrating that EID-1 strongly inhibited the transcriptional activity of SF-1 [27]. It was tempting to speculate that duplication of EID-1 may also be responsible for the hypogonadism phenotype.
Finally, we identified the SEMA6D gene as another candidate gene. SEMA6D gene, one member of the class VI subgroup of the semaphorin family, was expressed abundantly in kidney, brain, and placenta and moderately in the heart and skeletal muscles. The expression of SEMA6D predominantly in the adult tissues made them potentially important molecules in nervous system maintenance and repair. This profile was consistent with a more general role of the proteins in neurogenesis and organogenesis as well as in regenerative and degenerative processes [28]. Therefore, we hypothesized that overexpression of SEMA6D may play a role in neurodevelopmental anomalies of our patient.
In conclusion, we reported a de novo duplication of 15q15.3q21.2 in a patient with skeletal problems, hypogonadism, global developmental delay, intellectual disability and facial dysmorphism. Several candidate genes responsible for the complex clinical features were identified and critical region centered on 15q21.1 was defined as well, thus we proposed a novel contiguous gene duplication syndrome at this genomic interval. Further studies on duplications involving this region will be necessary.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med GenetA. 2008;146A(16):2041–52.
Battaglia A. The inv dup (15) or idic (15) syndrome (Tetrasomy 15q). Orphanet J Rare Dis. 2008;3:30.
Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, et al. The interstitial duplication 15q11.2–q13 syndrome includes autism mild facial anomalies and a characteristic EEG signature. Autism Res. 2013;6(4):268–79.
Lowther C, Costain G, Stavropoulos DJ, Melvin R, Silversides CK, Andrade DM, et al. Delineating the15q13.3 microdeletion phenotype: a case series and comprehensive review of the literature. Genet Med. 2015;17(2):149–57.
Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, Mahadevan S, et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum Mutat. 2010;31(7):840–50.
Tatton-Brown K, Pilz DT, Orstavik KH, Patton M, Barber JC, Collinson MN, et al. 15q overgrowth syndrome: a newly recognized phenotype associated with overgrowth, learning difficulties, characteristic facial appearance, renal anomalies and increased dosage of distal chromosome 15q. Am J Med Genet A. 2009;149A(2):147–54.
Poot M, Verrijn Stuart AA, van Daalen E, van Iperen A, van Binsbergen E, Hochstenbach R. Variable behavioural phenotypes of patients with monosomies of 15q26 and a review of 16 cases. Eur J Med Genet. 2013;56(7):346–50.
Palumbo O, Palumbo P, Palladino T, Stallone R, Miroballo M, Piemontese MR, et al. An emerging phenotype of interstitial 15q25.2 microdeletions: clinical report and review. Am J Med Genet A. 2012;158A(12):3182–9.
Wat MJ, Enciso VB, Wiszniewski W, Resnick T, Bader P, Roeder ER, et al. Recurrent microdeletions of 15q25.2 are associated with increased risk of congenital diaphragmatic hernia, cognitive deficits and possibly Diamond–Blackfan anaemia. J Med Genet. 2010;47(11):777–81.
Burgess T, Brown NJ, Stark Z, Bruno DL, Oertel R, Chong B, et al. Characterization of core clinical phenotypes associated with recurrent proximal 15q25.2 microdeletions. Am J Med Genet A. 2014;164A(1):77–86.
Doelken SC, Seeger K, Hundsdoerfer P, Weber-Ferro W, Klopocki E, Graul-Neumann L. Proximal and distal 15q25.2 microdeletions-genotype-phenotype delineation of two neurodevelopmental susceptibility loci. Am J Med Genet A. 2013;161A(1):218–24.
Elcioglu N, Fear C, Berry AC. Partial trisomy of 15q due to inserted inverted duplication. Clin Genet. 1997;52(6):442–5.
Herr HM, Scott Jr CI, Horton SJ. De novo interstitial direct duplication of 15q: 46, XY, dir dup(15) (pter leads to q24::q14 leads to q21 X 1::q24 leads to qter). J Med Genet. 1983;20(6):473–5.
Le Goff C, Cormier-Daire V. Genetic and molecular aspects of acromelic dysplasia. Pediatr Endocrinol Rev. 2009;6(3):418–23.
Tsilou E, MacDonald IM. Weill-Marchesani syndrome. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015.
Radonic T. Marfan syndrome. Ned Tijdschr Tandheelkd. 2013;120(12):665–8.
Muzza M, Rabbiosi S, Vigone MC, Zamproni I, Cirello V, Maffini MA, et al. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J Clin Endocrinol Metab. 2014;99(3):E544–53.
Yoshizawa-Ogasawara A, Ogikubo S, Satoh M, Narumi S, Hasegawa T. Congenital hypothyroidism caused by a novel mutation of the dual oxidase 2 (DUOX2) gene. J Pediatr Endocrinol Metab. 2013;26(1-2):45–52.
Carvalho DP, Dupuy C. Role of the NADPH Oxidases DUOX and NOX4 in Thyroid Oxidative Stress. Eur Thyroid J. 2013;2(3):160–7.
Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S, et al. Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab. 2008;93(2):605–10.
MacLellan WR, Xiao G, Abdellatif M, Schneider MD. A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol. 2000;20(23):8903–15.
Miyake S, Sellers WR, Safran M, Li X, Zhao W, Grossman SR, et al. Cells degrade a novel inhibitor ofdifferentiation with E1A-like properties upon exiting the cell cycle. Mol Cell Biol. 2000;20(23):8889–902.
Liu R, Lei JX, Luo C, Lan X, Chi L, Deng P, et al. Increased EID1 nuclear translocation impairs synapticplasticity and memory function associated with pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2012;45(3):902–12.
Hutchinson DT, Sullivan R. Rubinstein-Taybi Syndrome. J Hand Surg Am. 2015;40(8):1711–2.
Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab. 2015;29(4):607–19.
El-Khairi R, Achermann JC. Steroidogenic factor-1 and human disease. Semin Reprod Med. 2012;30(5):374–81.
Park YY, Park KC, Shong M, Lee SJ, Lee YH, Choi HS. EID-1 interacts with orphan nuclear receptor SF-1 and represses its transactivation. Mol Cells. 2007;24(3):372–7.
Qu X, Wei H, Zhai Y, Que H, Chen Q, Tang F, et al. Identification, characterization, and functional study of the two novel human members of the semaphoring gene family. J Biol Chem. 2002;277(38):35574–85.
Acknowledgments
This study makes use of data generated by the DECIPHER Consortium. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. We would like to thank the family of the proband for their cooperation with this study. We cherish our sincerest gratitude for Dr. Yiping Shen’s guidance and selfless help who is employed by Boston Children’s Hospital.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
HY carried out the cytogenetic studies and wrote the manuscript. MC carried out the cytogenetic studies. LL made the clinical evaluation and collected clinical information of the patient in detail. Others coordinated the clinical evaluation. All the authors have read and approved the manuscript.
Additional file
Additional file 1: Table S1.
qPCR confirmation of CMA results. (DOCX 22 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yuan, H., Meng, Z., Zhang, L. et al. A rare de novo interstitial duplication of 15q15.3q21.2 in a boy with severe short stature, hypogonadism, global developmental delay and intellectual disability. Mol Cytogenet 9, 2 (2016). https://doi.org/10.1186/s13039-016-0214-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-016-0214-3