
Abstract
Background
3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency (HMGCLD) is an autosomal recessive disorder of ketogenesis and leucine degradation due to mutations in HMGCL.
Method
We performed a systematic literature search to identify all published cases. Two hundred eleven patients of whom relevant clinical data were available were included in this analysis. Clinical course, biochemical findings and mutation data are highlighted and discussed. An overview on all published HMGCL variants is provided.
Results
More than 95% of patients presented with acute metabolic decompensation. Most patients manifested within the first year of life, 42.4% already neonatally. Very few individuals remained asymptomatic. The neurologic long-term outcome was favorable with 62.6% of patients showing normal development.
Conclusion
This comprehensive data analysis provides a systematic overview on all published cases with HMGCLD including a list of all known HMGCL mutations.
Similar content being viewed by others
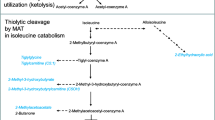


Background
The mitochondrial enzyme 3-hydroxy-3-methylglutaryl-coenzyme A lyase (HMGCL; EC 4.1.3.4) is required not only for the catabolism of the essential branched-chain amino acid leucine, but also for the synthesis of the ketone bodies acetoacetate and 3-hydroxy-n-butyrate [1]. Ketone bodies are an important source of energy for extrahepatic organs, in particular of the brain, in times of insufficient energy supply. Consequently, episodes of hypoglycemia and metabolic acidosis are an important observation in HMGCL deficiency (HMGLD; MIM246450). Due to the accumulation of characteristic leucine metabolites HMGCLD can be diagnosed via urinary organic acid analysis and usually is associated with an abnormal blood acylcarnitine profile as well. Confirmatory testing is available by enzyme activity assays in patient cells and by mutation analysis of the HMGCL gene. Recently, the number of individuals with confirmed HMGCLD has been estimated to be approximately 200 world-wide [1], but most information published so far is from case reports and small retrospective case series. Few studies have presented larger patient cohorts [2,3,4,5,6,7,8]. Although many patients reported originate from the Iberic peninsula and from Saudi Arabia, where HMGCLD is the most prevalent organic aciduria, HMGCLD is a panethnic disease. However, a comprehensive meta-analysis that covers all HMGCLD patients described in the literature so far is missing.
This has prompted us to approach a systematic assessment of all described patients with this ketogenesis defect.
Methods
We performed a systematic literature search in PubMed using the terms “3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency”, “3-Hydroxy-3-methylglutaryl-coA lyase deficiency”, “HMGCL deficiency”, and “3-HMG-coenzyme A lyase deficiency” in order to obtain information on the clinical course of all published patients. The search was performed in September 2019 and was completed by searches in the Human Gene Mutation Database (HGMD®) http://www.hgmd.cf.ac.uk/. This was supplemented by patient data from literature known to the authors due to their long-time work in the area of ketone body metabolism. All patients with metabolically, enzymatically and/or genetically proven HMGCLD on whom relevant clinical information was provided in the respective publication(s) were included in this study. With this approach we identified a total of 211 HMGCLD patients, mainly published in case reports as well as few case series. All cases were evaluated and analysed with a special focus on the patients’ age at onset, number of metabolic decompensations, clinical course including neurological outcome, treatment, residual enzyme activity and mutations in the HMGCL gene.
For the systematic overview on all published HMGCL mutations, publications that contained no clinical information were included in addition. A list of all publications included in the clinical data analysis is given in Additional file 1: Table S1, a list of publications that were additionally reviewed for mutations is provided in Additional file 2: Table S2.
Accuracy of age data ranged from hours to years in the different reports. For the calculation of median ages years were converted to months which might lead to an underestimation (i.e. 7 years = 84 months, although the patient might have been 7 years and 11 months old). In very few cases where only “newborn” was given for the age at report, we used 5 days of age for the calculation. If hours were given (“first symptoms 2 hours after birth”) these were rounded to days.
Results
Two hundred eleven cases of HMGCLD were identified and reviewed (Table 1, Additional file 1: Table S1). Seventy-eight patients were female, 101 were male, and the sex of the remaining 32 patients was not reported. The dataset included 8 pairs of siblings, thereof one pair of dizygotic twins. The age at last reported clinical follow-up was provided for 155 patients and ranged from 72 h to 40 years (median of 48 months). Forty-six patients were of Turkish origin, 20 Portuguese, 13 Brazilian of Portuguese ancestry, 19 Saudi Arabian, and 12 Spanish. All other origins accounted for 7 or less patients, and of 20 patients no ethnic or geographic origin was reported. Information on parental consanguinity was given in 122 cases with a consanguinity rate of 49% (60/122). 169 (80.1%) patients were alive at the time of report, 34 (16.1%) patients had deceased, and of 8 patients the outcome is not reported. Age at death ranged from 72 h to 40 years (median 9.5 months, n = 26). Most patients died due to metabolic decompensations, one of them at the age of 24 years during her second pregnancy [9]. One child deceased in his sleep at age 13 months with no apparent previous symptoms. One previously asymptomatic 29-year-old patient died of septic shock with multi-organic failure [10], one 7 month-old child due to cardiomyopathy and arrhythmias [11].
Information on the number of metabolic decompensations was available for 171 patients. Thereof, 163 patients (95.3%) suffered at least one metabolic crisis. In 8 patients more than 10 acute episodes were reported. Eight patients never had metabolic decompensations. Two of these patients were diagnosed asymptomatically by family screening [12, 13], three presented with seizures and/or developmental delay [2, 14,15,16] and two were diagnosed due to hepatomegaly and elevated plasma/serum activities of transaminases [2, 5]. One patient presented with macrocephaly, which was first noted at 2 months, as well as a doll-like facies with frontal bossing and depressed nasal bridge [17]. She also displayed a slight “sun-setting” phenomenon, tendency to opisthotonus and global developmental delay. Although this child never had a metabolic crisis a tendency towards hypoglycemia was reported [17,18,19,20]. One patient was diagnosed presymptomatically through family screening, but developed an acute decompensation in the third year of life [12].
Of 165 patients with acute symptoms the age at presentation was reported (146 cases with exact numbers, and 20 with some information, such as “neonatal onset” or “presentation in the third year of life). Median age at disease onset was 4 months (n = 146). 70/165 (42.4%) patients presented neonatally, 65 (39.4%) and 13 (7.9%) in the first and second year of life, respectively, while the remainder of 17 patients (17/165; 10.8%) showed first symptoms beyond the second year of life only (Fig. 1). Within the neonatal onset group 11 patients were already symptomatic on the first day of life. The latest manifestation was reported in a 29-year-old patient who died during the initial metabolic crisis due to multiorgan failure [10]. There was often a significant delay until the correct diagnosis could be made. In one patient it took 36 years between the onset of symptoms and the time of diagnosis [2, 21].

Age at first presentation of 165 HMGCLD patients with acute symptoms. The vast majority of patients presented within the first year of life with neonatal onset in more than 40% of patients. The latest manifestation was observed at 29 years
Clinical symptoms of acute decompensations mainly comprised vomiting, lethargy/ coma, tachypnea/apnoea, seizures and moderate hepatomegaly. Few patients presented with stroke-like episodes. Common laboratory findings were (severe) hypoglycemia, metabolic acidosis, elevated activities of serum transaminases and hyperammonemia. Transaminase activities were often only mildly increased, but episodes of transient elevations up to > 1000 U/l have been reported in few patients [22, 23]. One child developed an episode of liver dysfunction with massively elevated transaminase activities (AST 4150 IU/L, ALT 2200 IU/L) at age 5 months [22], and another patient showed an AST activity of 11,736 IU/l during a severe metabolic decompensation at age 7 months [23]. Hyperammonemia was rather mild in most cases, however ammonia levels > 1000 μmol/L have been described, and one patient even presented with severe hyperammonemia of > 2000 μmol/l requiring peritoneal dialysis [24].
Information on the neurologic outcome was available on 140 patients (Fig. 2). Thereof, 87 (87/139; 62.6%) showed normal psychomotor development without neurologic abnormalities. One 2-year-old patient had trisomy 21 [25] and was therefore not included in the analysis. In 9 patients (9/139; 6.5%) only slight abnormalities were reported including muscular hypotonia or a transiently increased muscle tone, hyperactivity and partial performance weaknesses such as dyslexia and difficulties in grammar. Forty-four patients (44/139; 31.7%) showed developmental delay or distinct neurologic abnormalities. Eighteen patients were described as severely retarded, 6 had a moderate and 4 a mild disability. In 5 cases the degree of neurologic impairment was not further specified. Ten patients showed neurologic symptoms including spastic hemiparesis or tetraplegia, distinct muscular hypotonia, impairment of vision and hearing, cerebellar ataxia, movement disorders, tremor, clonic movements, mild dysarthria, exaggerated deep tendon reflexes and absence of social contact. Seizures were reported in 13 patients (9.0%).

Cognitive development and neurologic complications in 139 HMGCLD patients. 62.6% of patients show normal development, while severe mental disability is rather rare in this patient cohort. Neurologic symptoms were documented in 10 patients including spastic hemiparesis or tetraplegia, distinct muscular hypotonia, impairment of vision and hearing, cerebellar ataxia, movement disorders, tremor, clonic movements, mild dysarthria, exaggerated deep tendon reflexes and absence of social contact. Seizures were reported in 13 patients
Imaging data (MRI or CT) were available of 60 patients. There will of course be a bias in favour of abnormal findings as imaging is primarily performed in patients with neurologic symptoms. Nevertheless, it is notable that imaging results were unremarkable in only 2 children [2, 26]. The most common findings were white matter changes which were uniformly present in almost all patients. Another frequent observation was cerebral atrophy with dilatation of the ventricular system. Abnormalities reported in a few or single patients comprised basal ganglia involvement, demyelinisation, ischemic lesions, chronic subdural hematoma, subdural hygroma and bilateral occipital porencephaly.
In 117 cases information on dietary regimens is available. Nine of these patients (7.7%) had no dietary restrictions, although in one of them a low-leucine diet was recommended. The remaining 108 patients followed a specific diet at least temporarily. Forty-six patients (46/105; 43.8%) were on either a low-leucine (28 patients) or low-protein (18 patients) diet, 57 patients (57/105; 54.3%) followed a diet low in leucine/protein and fat. Only one patient had a fat-restricted diet without protein restriction (1/105; 1%). Few patients had a self-imposed diet already before diagnosis [13, 27,28,29]. In three cases it was only stated that a diet was given, but no details were provided. Many patients on leucine/ protein restriction received supplementation with a leucine-free amino acid mixture. Some patients received additional carbohydrate supplementation either by corn starch or by glucose polymers. Avoidance of fasting was usually recommended. One patient received long-term treatment with diazoxide (25 mg/8 h) [20]. Some patients were supplemented with bicarbonate. In some patients the diet was relaxed at some point during childhood. For 109 patients, data on carnitine treatment was available. In this cohort, carnitine was supplemented in 85 cases (78%), while 24 (22%) patients received no carnitine supplementation.
Apart from neurologic symptoms long-term complications affecting other organs seem to be rather rare. Three patients developed dilated cardiomyopathy with arrhythmias which were fatal in two cases [2, 11, 30], and in one patient left ventricular noncompaction was diagnosed [31]. Two patients were reported with pancreatitis, one 5-year-old girl with a single episode [31] and one boy with recurrent episodes [22].
In 4 of 216 cases, HMCGL deficiency was reported in association with another congenital disorder. One patient had a trisomy 21 [25], one patient was reported with VATERL syndrome [32], and one patient had a situs inversus totalis and gastroschisis [33]. In the fourth patient who presented with deafness and retinitis pigmentosa an Usher syndrome type I, a rare autosomal recessive condition of profound congenital deafness and severe retinitis pigmentosa associated with developmental delay, was suspected [27].
A total of 8 pregnancies have been reported in 5 women [9, 34,35,36]. Five pregnancies resulted in healthy offspring, while one mother who already presented with recurrent metabolic decompensations during her first pregnancy died during her second pregnancy at 9 weeks of gestation due to maternal metabolic decompensation [9]. In one patient intrauterine death occurred in the first pregnancy at 10 weeks of gestation during maternal metabolic decompensation, and the second pregnancy was terminated at 6 weeks of gestation in absence of metabolic problems [9].
Enzymatic studies have been performed in 114 patients confirming a reduced or absent HMGCL activity in leukocytes, Epstein-Barr virus (EBV)-transformed lymphoblastoid cells or fibroblasts in all of them.
Results of HMGCL mutation analysis were reported for 118 patients. Mutations were identified in all 9 exons of HMGCL and also in noncoding regions of the gene. An overview on all mutations identified in the HMCGL gene that were reported in the literature so far is given in Fig. 3 (following transcription into the current nomenclature, where required). Eighty-six patients (72.9%) carried homozygous mutations and 24 patients (20.3%) were compound heterozygous for variants in the HMGCL gene. In one of the homozygous patients, paternal uniparental isodisomy of chromosome 1 was confirmed (Aoyama 2015). In 6 patients (5.1%) only one mutation was detected, and in 2 patients (1.7%) no mutation could be identified although HMGCL activity was clearly deficient in fibroblasts [2, 37]. The most common HMGCL mutation was the c.109G > T, p.(Glu37*) variant that was found in a total of 36 patients (30.5%), mostly in homozygosity (homozygous in 28 patients, heterozygous in 8 patients). It was mostly reported in individuals originating from the geographically/ demographically linked countries Brazil, Spain, Portugal and Morocco, but also in two Pakistani patients. The two other common variants that were identified in 12 (10.2%) and 6 patients (5.5%), respectively, were c.122G > A, p.(Arg41Gln) and the splice site mutation c.876 + 1G > C. While p.(Arg41Gln) was mostly found in individuals from Saudi Arabia, it was also noted in individuals of Turkish and Italian origin. c.876 + 1G > C is common among Turkish patients with HMGCLD. All other mutations were reported in less than 5 individuals.

Mutations reported for human HMGCL. a: c.27del has been demonstrated by Pospísilová et al. 2003 to lead to a frame shift [39]. This mutation leads to a frameshift and premature stop codon after 32 amino acids without degradation of the DNA, while p.(Arg10Glyfs*24) would be predicted. b: associated with skipping of exon 2 [15]. c: Likely to affect splicing, although not proven [40]. d: Has been named p.Val168Valfs8 by Puisac et al. 2013; may also cause skipping of exon 5 or of exons 5 and 6 (the latter resulting in a physiological mRNA transcript according to [41]. e: According to Buesa et al. 1996 aberrant splicing, which mostly results in skipping of exon 9, p.(Met251_Thr292del,), but to a small extent in insertion of 17 amino acids, which precede a stop codon: p.(fs*18) [42]. Not displayed: -Pie et al. (1997) reported a 84 bp in-frame deletion on the mRNA level leads to the loss of 28 amino acids (Val-21 to Lys-48) in the mature protein [43]. This deleted region includes the last of the leader peptide of the precursor HL protein and 21 amino acids of the N-terminus of the mature protein. -Deletion (between intron 1 and intron 4) NG_013061: g.9326_13806del reported by Aoyama et al. 2015 [44]. - Mutation r.61-144del identified on the RNA level only [2]. - As the skipping of exons 5-6b skipping of exons 5–7 has been reported for a physiological alternative transcript [41]. Note: Zaferiou et al. 2007 referred to a ‘C-to-T transition’ which actually should be c.796C > T and is indicated as such in this figure [45]. Roland et al. 2017: c.438 T > G, p.(Ser46Arg) was corrected to p.(Ser146Arg) [37]
Discussion/conclusion
This work aims at a comprehensive overview on the clinical course, biochemical and genetic data of all patients with HMGCLD published in the literature so far.
Patients with HMGCLD typically present with acute metabolic decompensation that may be life-threatening. Very few patients were diagnosed with only chronic, mainly neurologic symptoms. Interestingly, only very few asymptomatic patients have been described although HMGCLD is a target disease of newborn screening programs in several countries. This may of course be due to a publication bias as asymptomatic individuals are often not reported, and some of the individuals identified by newborn screening were described in papers with no further relevant clinical information and therefore not included in this analysis.
Of the symptomatic patients 42.4% presented neonatally and more than 80% within the first year of life, while manifestation beyond the first year of life was the exception. This is compatible with the special role of ketone bodies for the energy supply of the newborn. Despite the often early and severe manifestation the long-term outcome seems to be favourable with the majority of patients showing normal cognitive development. Taking into consideration that our analysis also included patients that were diagnosed and treated as early as in the 1970s, the prognosis of patients born today may be even better than assumed based on this cohort.
As in other rare inherited metabolic diseases no controlled treatment studies are available for HMCGLD. Therefore, no conclusions can be drawn with respect to the necessity of a special diet or carnitine supplementation from our data although the majority of patients was on a protein and/or fat restricted diet. Based on pathobiochemical considerations and clinical reports the avoidance of fasting seems to be the mainstay of therapy in this disorder of ketogenesis. Administration of L-carnitine may have detoxifying effects and help to avoid secondary L-carnitine deficiency and intracellular depletion of free coenzyme A [38].
Our data demonstrate that HMGCLD is a panethnic disease, although some mutations are clustered in certain geographic areas with close connections throughout history. Interestingly, 18 patients carrying a homozygous mutation in HMGCL were explicitly reported to be the offspring of a non-consanguineous union. This possibly reflects an underestimation of parental consanguinity in this patient cohort. In line with previous reports on patient subgroups, our comprehensive study underlines that “genotype–phenotype correlations are difficult to establish” in HMGCLD [2, 6].
Conclusion
Despite its often early and severe manifestation HMGCLD seems to be associated with a favorable long-term outcome in the majority of cases.
Availability of data and materials
Raw data of this analysis are available on request.
Change history
10 January 2022
A Correction to this paper has been published: https://doi.org/10.1186/s13023-021-02154-z
Abbreviations
- HMGCL:
-
3-hydroxy-3-methylglutaryl coenzyme A lyase
- HMGCLD:
-
3-hydroxy-3-methylglutaryl coenzyme A lyase deficiency
References
Sass JO, Fukao T, Mitchell GA. Inborn errors of ketone body Metabolismand transport: an update for the clinic and for clinical laboratories. J Inborn Errors Metab Screen. 2018;6:1–7. https://doi.org/10.1177/2326409818771101.
Grünert SC, Schlatter SM, Schmitt RN, Gemperle-Britschgi C, Mrázová L, Balcı MC, et al. 3-Hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: clinical presentation and outcome in a series of 37 patients. Mol Genet Metab. 2017;121:206–15.
Cardoso ML, Rodrigues MR, Leão E, Martins E, Diogo L, Rodrigues E, et al. The E37X is a common HMGCL mutation in Portuguese patients with 3-hydroxy-3-methylglutaric CoA lyase deficiency. Mol Genet Metab. 2004;82:334–8.
Gibson KM, Breuer J, Nyhan WL. 3-Hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: review of 18 reported patients. Eur J Pediatr. 1988;148:180–6.
Menao S, López-Viñas E, Mir C, Puisac B, Gratacós E, Arnedo M, et al. Ten novel HMGCL mutations in 24 patients of different origin with 3-hydroxy-3-methyl-glutaric aciduria. Hum Mutat. 2009;30:E520–9.
Pié J, López-Viñas E, Puisac B, Menao S, Pié A, Casale C, et al. Molecular genetics of HMG-CoA lyase deficiency. Mol Genet Metab. 2007;92:198–209.
Vargas CR, Sitta A, Schmitt G, Ferreira GC, Cardoso ML, Coelho D, et al. Incidence of 3-hydroxy-3-methylglutaryl-coenzyme a lyase (HL) deficiency in Brazil, South America. J Inherit Metab Dis. 2008;31(Suppl 3):511–5.
Ozand PT, al Aqeel A, Gascon G, Brismar J, Thomas E, Gleispach H. 3-Hydroxy-3-methylglutaryl-coenzyme a (HMG-CoA) lyase deficiency in Saudi Arabia. J Inherit Metab Dis. 1991;14:174–88.
Langendonk JG, Roos JCP, Angus L, Williams M, Karstens FPJ, de Klerk JBC, et al. A series of pregnancies in women with inherited metabolic disease. J Inherit Metab Dis. 2012;35:419–24.
Reimão S, Morgado C, Almeida IT, Silva M, Corte Real H, Campos J. 3-Hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: initial presentation in a young adult. J Inherit Metab Dis. 2009;32(Suppl 1):S49–52.
Gibson KM, Cassidy SB, Seaver LH, Wanders RJ, Kennaway NG, Mitchell GA, et al. Fatal cardiomyopathy associated with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. J Inherit Metab Dis. 1994;17:291–4.
van der Knaap MS, Bakker HD, Valk J. MR imaging and proton spectroscopy in 3-hydroxy-3-methylglutaryl coenzyme a lyase deficiency. AJNR Am J Neuroradiol. 1998;19:378–82.
Bakker HD, Wanders RJ, Schutgens RB, Abeling NG, van Gennip AH. 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency: absence of clinical symptoms due to a self-imposed dietary fat and protein restriction. J Inherit Metab Dis. 1993;16:1061–2.
Casale CH, Casals N, Pié J, Zapater N, Pérez-Cerdá C, Merinero B, et al. A nonsense mutation in the exon 2 of the 3-hydroxy-3-methylglutaryl coenzyme a lyase (HL) gene producing three mature mRNAs is the main cause of 3-hydroxy-3-methylglutaric aciduria in European Mediterranean patients. Arch Biochem Biophys. 1998;349:129–37.
Puisac B, Teresa-Rodrigo ME, Arnedo M, Gil-Rodríguez MC, Pérez-Cerdá C, Ribes A, et al. Analysis of aberrant splicing and nonsense-mediated decay of the stop codon mutations c.109G>T and c.504_505delCT in 7 patients with HMG-CoA lyase deficiency. Mol Genet Metab. 2013;108:232–40.
Yýlmaz Y, Ozdemir N, Ekinci G, Baykal T, Kocaman C. Corticospinal tract involvement in a patient with 3-HMG coenzyme a lyase deficiency. Pediatr Neurol. 2006;35:139–41.
Lisson G, Leupold D, Bechinger D, Wallesch C. CT findings in a case of deficiency of 3-hydroxy-3-methylglutaryl-CoA-lyase. Neuroradiology. 1981;22:99–101.
Leupold D, Bojasch M, Jakobs C. 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in an infant with macrocephaly and mild metabolic acidosis. Eur J Pediatr. 1982;138:73–6.
Wysocki SJ, Hähnel R. 3-Hydroxy-3-methylglutaric aciduria: deficiency of 3-hydroxy-3-methylglutaryl coenzyme a lyase. Clin Chim Acta. 1976;71:349–51.
Gibson KM, Breuer J, Kaiser K, Nyhan WL, McCoy EE, Ferreira P, et al. 3-Hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: report of five new patients. J Inherit Metab Dis. 1988;11:76–87.
Bischof F, Nägele T, Wanders RJA, Trefz FK, Melms A. 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in an adult with leukoencephalopathy. Ann Neurol. 2004;56:727–30.
Muroi J, Yorifuji T, Uematsu A, Nakahata T. Cerebral infarction and pancreatitis: possible complications of patients with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. J Inherit Metab Dis. 2000;23:636–7.
Muroi J, Yorifuji T, Uematsu A, Shigematsu Y, Onigata K, Maruyama H, et al. Molecular and clinical analysis of Japanese patients with 3-hydroxy-3-methylglutaryl CoA lyase (HL) deficiency. Hum Genet. 2000;107:320–6.
Stacey TE, de Sousa C, Tracey BM, Whitelaw A, Mistry J, Timbrell P, et al. Dizygotic twins with 3-hydroxy-3-methylglutaric aciduria; unusual presentation, family studies and dietary management. Eur J Pediatr. 1985;144:177–81.
Ferreira G, Freitas S, Pereira SA, Martins I, Tavares E, Vilarinho L. 3-Hydroxy-3-methylglutaric aciduria in a girl with trisomy 21. Eur J Pediatr. 1996;155:1068.
Gibson KM, Lee CF, Kamali V, Johnston K, Beaudet AL, Craigen WJ, et al. 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency as detected by radiochemical assay in cell extracts by thin-layer chromatography, and identification of three new cases. Clin Chem. 1990;36:297–303.
Jones KJ, Wilcken B, Kilham H. The long-term evolution of a case of 3-hydroxy-3-methylglutaryl-coenzyme a lyase deficiency associated with deafness and retinitis pigmentosa. J Inherit Metab Dis. 1997;20:833–4.
Wysocki SJ, Hähnel R. 3-Hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: a review. J Inherit Metab Dis. 1986;9:225–33.
Shilkin R, Wilson G, Owles E. 3-Hydroxy-3-methylglutaryl coenzyme a lyase deficiency. Follow-up of first described case. Acta Paediatr Scand. 1981;70:265–8.
Leung AAC, Chan AK, Ezekowitz JA, Leung AKC. A case of dilated cardiomyopathy associated with 3-Hydroxy-3-Methylglutaryl-coenzyme a (HMG CoA) Lyase deficiency. Case Rep Med. 2009;2009:183125.
Köksal T, Gündüz M, Özaydın E, Azak E. 3-HMG coenzyme a Lyase deficiency: macrocephaly and left ventricular noncompaction with a novel mutation. Indian J Pediatr. 2015;82:645–8.
al-Essa M, Rashed M, Ozand PT. 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency in a boy with VATER association. J Inherit Metab Dis. 1998;21:443–4.
Kose M, Kagnıcı M, Canda E, Altinol Y, Ceylaner S, Aalkan K, et al. HMG-CoA lyase deficiency: one disease three pictures. J Inherit Metab Dis. 2014;37(Suppl 1):S107.
Pipitone A, Raval DB, Duis J, Vernon H, Martin R, Hamosh A, et al. The management of pregnancy and delivery in 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Am J Med Genet A. 2016;170:1600–2.
Sulaiman RA, Al-Nemer M, Khan R, Almasned M, Handoum BS, Al-Hassnan ZN. Successful Management of Pregnancies in patients with inherited disorders of ketone body metabolism. JIMD Rep. 2018;38:41–4.
Santosa D, Donner MG, Vom Dahl S, Fleisch M, Hoehn T, Mayatepek E, et al. Favourable outcome in two pregnancies in a patient with 3-Hydroxy-3-Methylglutaryl-CoA Lyase deficiency. JIMD Rep. 2017;37:1–5.
Roland D, Jissendi-Tchofo P, Briand G, Vamecq J, Fontaine M, Ultré V, et al. Coupled brain and urine spectroscopy - in vivo metabolomic characterization of HMG-CoA lyase deficiency in 5 patients. Mol Genet Metab. 2017;121:111–8.
Mitchell GA, Gauthier N, Lesimple A, Wang SP, Mamer O, Qureshi I. Hereditary and acquired diseases of acyl-coenzyme a metabolism. Mol Genet Metab. 2008;94:4–15.
Pospísilová E, Mrázová L, Hrdá J, Martincová O, Zeman J. Biochemical and molecular analyses in three patients with 3-hydroxy-3-methylglutaric aciduria. J Inherit Metab Dis. 2003;26:433–41.
Al-Sayed M, Imtiaz F, Alsmadi OA, Rashed MS, Meyer BF. Mutations underlying 3-hydroxy-3-methylglutaryl CoA lyase deficiency in the Saudi population. BMC Med Genet. 2006;7:86.
Puisac B, Ramos M, Arnedo M, Menao S, Gil-Rodríguez MC, Teresa-Rodrigo ME, et al. Characterization of splice variants of the genes encoding human mitochondrial HMG-CoA lyase and HMG-CoA synthase, the main enzymes of the ketogenesis pathway. Mol Biol Rep. 2012;39:4777–85.
Buesa C, Pié J, Barceló A, Casals N, Mascaró C, Casale CH, et al. Aberrantly spliced mRNAs of the 3-hydroxy-3-methylglutaryl coenzyme a lyase (HL) gene with a donor splice-site point mutation produce hereditary HL deficiency. J Lipid Res. 1996;37:2420–32.
Pié J, Casals N, Casale CH, Buesa C, Mascaró C, Barceló A, et al. A nonsense mutation in the 3-hydroxy-3-methylglutaryl-CoA lyase gene produces exon skipping in two patients of different origin with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Biochem J. 1997;323(Pt 2):329–35.
Aoyama Y, Yamamoto T, Sakaguchi N, Ishige M, Tanaka T, Ichihara T, et al. Application of multiplex ligation-dependent probe amplification, and identification of a heterozygous Alu-associated deletion and a uniparental disomy of chromosome 1 in two patients with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Int J Mol Med. 2015;35:1554–60.
Zafeiriou DI, Vargiami E, Mayapetek E, Augoustidou-Savvopoulou P, Mitchell GA. 3-Hydroxy-3-methylglutaryl coenzyme a lyase deficiency with reversible white matter changes after treatment. Pediatr Neurol. 2007;37:47–50.
Acknowledgements
We are grateful to Albert VI, Archduke of Austria, for founding the University of Freiburg in 1457, and to the German State of North Rhine-Westphalia for founding Bonn-Rhein-Sieg University of Applied Sciences in 1995.
Funding
J.O. Sass gratefully acknowledges financial support by the program ‘FH Zeit für Forschung’ (project ‘KETOplus’, 005–1703-0016) of the Ministry of Culture and Science of the German State of North Rhine-Westphalia.
Author information
Authors and Affiliations
Contributions
SCG performed the systematic literature search and JOS the search and renaming of mutations. Both authors contributed additional literature and drafted parts of the manuscript, revised the other parts and agreed to all contents.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
As this study is a retrospective analysis of data published in the literature already no ethic approval was required.
Consent for publication
Not applicable.
Competing interests
SCG declares that she does not have competing interests. JOS serves as consultant of a medical diagnostic laboratory in ketone body metabolism and he and his institution have received honoraria/a reimbursement by Swedish Orphan Biovitrum GmbH.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Publications included in this literature review for the analysis of clinical, biochemical and genetic data.
Additional file 2: Table S2.
Publications that were additionally included for the overview on all known mutations in HMGCL.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Grünert, S.C., Sass, J.O. 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: one disease - many faces. Orphanet J Rare Dis 15, 48 (2020). https://doi.org/10.1186/s13023-020-1319-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-1319-7