
Abstract
Background
Laing distal myopathy is a rare autosomal dominant inherited distal myopathy caused by mutations of the MYH7 gene affecting mainly the rod region. We described the clinical features, muscle MRI and pathological changes as well as genetic mutations in a group of Chinese patients with Laing distal myopathy.
Results
Six patients with the confirmed diagnoses of Laing distal myopathy were recruited. Ankle dorsiflexion and finger extension weakness, as well as neck flexion weakness were common in our patients. Myopathic as well as neurogenic lesions were suggested by electromyography in different patients. Respiratory abnormality of sleep apnea was detected in two of our patients stressing the necessity of close respiratory monitoring in this disease. Muscle MRIs showed similar features of concentric fatty infiltration of anterior thigh muscles together with early involvement of tibialis anterior and extensor hallucis longus. However, muscle pathological presentations were varied depending on the biopsied muscles and the severity of the disease. In-frame deletions of the MYH7 gene made up 3/4 of mutations in our patients, suggesting that these are common mutations of Laing distal myopathy.
Conclusions
Our study further expanded the phenotypes and genotypes of Laing distal myopathy. In-frame deletions of the MYH7 gene are common causes of Laing distal myopathy.
Similar content being viewed by others
Background
Myosin is a family of highly conserved proteins acting as a molecular motor, which provides mechanical forces in a variety of cellular movements including muscle contraction [1]. Muscle myosin is myosin II consisting of two myosin heavy chain (MyHC) subunits and two pairs of light chain subunits [2]. There are several MyHC isoforms of striated muscles which are expressed in different muscle types. Among these, slow/β-cardiac MyHC is encoded by MYH7, and is mainly expressed in slow, type 1 muscle fibers as well as in heart ventricles [3].
Slow/β-cardiac MyHC can be divided into two parts: the N-terminal globular head region made by amino acids 1-847, and the C-terminal rod region made by amino acids 848-1935 [4]. A variety of both cardiac and skeletal muscle disorders can be caused by MYH7 mutations. Mutations affecting the globular head region mainly cause cardiomyopathies, including familial hypertrophic/dilated cardiomyopathy, and left ventricular non-compaction (LVNC) cardiomyopathy, while mutations affecting the rod region mainly cause skeletal myopathies, including myosin storage myopathy (MSM), Laing distal myopathy (LDM) [5].
Laing distal myopathy is inherited as autosomal dominant with typical features of early-onset distal weakness mainly affecting ankle dorsiflexors and finger extensors, resulting in a “hanging big toe” sign. Neck flexion weakness is also very common in LDM. The disease course is usually very slow, with most patients remaining ambulant. Cardiac involvement is seldom seen in LDM, and serum creatine kinase (CK) levels are usually normal or only moderately elevated [6, 7]. Muscle MRI of LDM often shows the tibialis anterior as the earliest and most severely affected muscles [8]. However, no consistent conclusions of other muscles involvement on MRI are available. Muscle pathological changes in LDM are also variable and unspecific. Common pathological findings include fiber size variation, abnormally small type 1 fibers, type 1 or 2 fiber predominance, internal nuclei, minicores, and mitochondrial abnormalities [9].
Up to now, a number of both sporadic and familial LDM cases have been reported around the world, including several Chinese cases [10,11,12]. Here, we report three Chines LDM pedigrees and one sporadic Chinese LDM patient, and describe the clinical, radiographical, pathological and genetic features of these cases, to further expand both the genotypes and the phenotypes of LDM.
Materials and methods
Patients
We retrospectively reviewed patients with muscular disorders from Peking University First Hospital, and recruited patients suspected of Laing distal myopathy. All recorded clinical information was collected, including medical histories of clinical symptoms, distributions of muscle weakness, cardiac and pulmonary symptoms or evaluations, serum CK levels, and electromyography findings et al. Muscle strengths were evaluated with the Medical Research Council (MRC) scale. This study was approved by the Human Research Ethics Committee of Peking University First Hospital, and all participants provided written informed consent.
Muscle MRI
Muscle MRIs using 1.5-T or 3.0-T magnetic resonance scanners (GE) of the thighs and/ or the calves were performed in five patients. Axial scanning in conventional T1-weighted and short T1 inversion recovery (STIR) sequences were carried out and additional images were acquired in the coronal plane when necessary. The detailed protocol for the scanning was according to our previous study [13]. The MRI findings were interpreted by an experienced radiologist and a neurologist independently and any disagreements were resolved by consensus.
Muscle biopsy
Four patients received muscle biopsies. Muscle specimens were obtained from biceps in three patients, and from tibialis anterior in one patient. The muscle specimens were frozen in isopentane, and cooled in liquid nitrogen. Serial frozen sections were stained by routine histological and histochemical methods and by standard immunohistochemical techniques for dystrophin (N-terminus, C-terminus, and rod domain, Novocastra), sarcoglycan complex, (α-, β-, and γ-, Novocastra), dysferlin (Chemicon), and desmin (Novocastra).
Genetic tests
Genomic DNA was extracted from peripheral blood samples of all enrolled patients and their available family members using standard procedures. Variants were detected by next-generation sequencing using the same protocol as previously described [14]. Sanger sequencing with specific primers was performed to confirm the selected variants. In addition, all the available family members of the patients were tested to confirm the segregation.
Results
Patients and clinical features
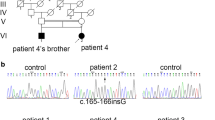
Five familial patients from three pedigrees and one sporadic patient with the diagnoses of Laing distal myopathy were recruited (Fig. 1). All six patients underwent routine cardiac and respiratory examinations, including electrocardiogram, Holter, echocardiography and pulmonary function test. Detailed clinical features were summarized in Table 1.

Enrolled familial and sporadic cases in the study
Patient II-6 and III-1 were from a large three-generation family A. Patient II-6 presented with abnormal toe walking at the age of 20, and found difficulty in climbing stairs at 31, then gradually showed finger weakness. She developed weakness in neck flexion and winging scapula at the age of 48. Examination at the age of 48 revealed significant decrease of muscle strengths in neck flexion (2/5), finger extension (4−/5), ankle dorsiflexion (0/5) and ankle plantarflexion (3 + /5), and mild decrease of proximal limb strengths (4/5 on upper limbs and 4−/5 on lower limbs) with all deep tendon reflexes absent. Contractures of Achilles tendons and winging scapula were present. No cardiac symptoms or abnormal examinations were found but with symptoms of sleep apnea. Serum CK was normal and EMG showed features of myogenic lesions. Patient III-1 was her 13-year-old son who developed bilateral Achilles tendon contracture since 8 years old. Strength tests only showed slight decrease in neck flexion, ankle dorsiflexion and proximal lower limbs. The rest family members were not affected.
Patient II-2 and III-1 were from a small three-generation family B. Father of patient II-2 was found abnormal gait since middle age but had not been examined or diagnosed. Patient II-2 only presented with slight symptoms of hand and foot weakness which did not affect her daily life. Examination showed slight decrease of muscle strengths in neck flexion (4/5), finger extension (4/5) and ankle dorsiflexion (4/5). No orthopedic, cardiac or respiratory abnormalities were found, and serum CK was also normal. Her daughter, patient III-1, presented with bilateral finger extension weakness since childhood, which progressed very slowly as she came to the clinic at the age of 20. Examination showed moderate decrease of muscle strengths in finger extension (3/5), and slight decrease of muscle strengths in neck flexion (4 + /5) and ankle dorsiflexion (4−/5). Scoliosis and talipes cavus were also present. Neither cardiac nor respiratory abnormalities were found. Serum CK was moderately elevated. EMG suggested neurogenic lesions with spontaneous activities as well as large motor unit potentials in several muscles, but nerve conduction studies were normal.
Patient III-1 was also from a three-generation family C. His grandfather was said to be weak in both legs while standing but was able to walk and climb stairs. His father presented with strephenopodia and abnormal gait since young. But neither of them was examined by clinicians. The onset symptoms of patient III-1 were strephenopodia and abnormal gait at the age of 7, and then he received orthopedic surgery. Examination showed slight decrease of muscle strengths in neck flexion (4/5), proximal upper limbs (4/5), finger extension (4/5) and ankle dorsiflexion (4-/5) with all deep tendon reflexes absent. Contracture of Achilles tendon, scoliosis and winging scapula were also present. Sleep apnea was obvious since 18 years old, so he had to use non-invasive positive pressure ventilation during sleep. No cardiac abnormalities were found, and serum CK was normal. EMG showed neurogenic lesions with spontaneous activities and large motor unit potentials in most of the muscles tested, but with normal nerve conduction studies.
Patient II-2 was the only symptomatic one in her family D, who could not run as fast as peers during childhood. After tug-of-war during high school, she was unable to lift her left arm. Examination at the age of 29 showed normal strength of neck flexion, but 0/5 in her left upper arm with normal strength in her right proximal arm. Muscle strengths of both fingers extension and ankles dorsiflexion were 0/5. No orthopedic, cardiac or respiratory abnormalities were found, and serum CK was normal. EMG showed myogenic lesions.
Muscle imaging
Thigh and calf muscle MRIs were performed in all enrolled patients except patient III-1 (family A). The distributions of fatty infiltration were listed in Table 2 and shown in Fig. 2, which were not uniform among different patients. For thigh muscles, vastus intermedius and adductor magnus were severely affected in all the examined patients except patient III-1 (family C), in whom all thigh muscles were with diffusely mildly fatty infiltration. Other thigh muscles severely affected included semitendinosus in three patients, sartorius in two, semitendinosus in two, adductor longus in one and gracilis in one. Rectus femoris was relatively preserved in all examined patients, while other muscles were affected differently. For calf muscles, tibialis anterior and extensor hallucis longus were severely affected in all patients, while soleus was severely affected in two. Other calf muscles were mildly affected or relatively preserved. Patient II-2 (family D) also did lumbar MRI, which showed obvious fatty infiltration of paraspinal and other axial muscles (Fig. 2).

Muscle MRIs of the patients. a–j are lower muscle MRIs, each row belongs to one patient which are patient II-6 (family A), II-2 (family B), III-1 (family B), III-1 (family C) and II-2 (family D) respectively. Thigh muscle MRIs are on the left, and calf muscle MRIs are on the right. k–l are lumbar muscle MRIs of patient II-2 (family D). Detailed descriptions of the MRIs are present in the main text
Muscle pathology
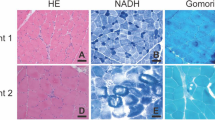
Muscle biopsies were done in patient II-6 (family A), II-2 (family B), II-2 (family D) on biceps and in patient III-1 (family C) on tibialis anterior. Pathological changes were summarized in Table 3. Increased fiber size variation was seen in all patients, while other common findings included type 2 fiber predominance in patient II-6 (family A), II-2 (family B) and III-1 (family C), nuclei internalization in patient II-6 (family A) and III-1 (family C). Specific findings included fiber type grouping and subsarcolemmal abnormal mitochondrial in patient III-1 (family C), as well as multi-cores and small type 1 fibers in patient II-2 (family D) (Fig. 3).

Muscle pathological changes of the patients. a–c are ATPase staining at pH 10.6, 10.7 and 10.6 of patient II-6 (family A), II-2 (family B) and III-1 (family C) respectively, showing type 2 fibers predominance. c also shows fiber type grouping. d is H&E staining of patient III-1 (family C) showing increased fiber size variation, necrotic and degenerated fibers. e is SDH staining of patient III-1 (family C) showing subsarcolemmal mitochondria accumulation. F is NADH-TR staining of patient II-2 (family D) showing multiple cores. All bar = 200 μm. NADH-TR, nicotinamide adenine dinucleotide tetrazolium reductase; SDH, succinate dehydrogenase
Genetic findings
Mutations of MYH7 were detected in all patients and not detected in non-affected family members (Table 3). Deletions of three nucleotides in exon 33 or 34 causing deletion of one amino acid were detected in family A, C and D. A missense mutation in exon 35 was detected in family B. Mutations in family A, B, and D had been reported as pathogenic in previous researches [5, 8, 15, 16]. The deletion mutation in family C was regarded as likely pathogenic according to ACMG guidelines.
Discussion
MYH7 mutations can cause two main groups of myopathies including myosin storage myopathy and Laing distal myopathy. In this study, we have described a series of patients with genetic confirmed Laing distal myopathy. Up to now, there have been more than 30 articles on Laing distal myopathy, including three reports on Chinese patients [10,11,12]. So far, this is the largest report on Laing distal myopathy in China. The onset ages are below 10 years old in young patients, but are 20–30 years old in middle-aged patients. This might be explained by the less awareness of weakness in patients or due to the relatively wide range of onset ages of this disease [9, 17]. The clinical features of this disease are characterized by slowly progressive muscle weakness of ankle dorsiflexion and finger extension [9]. The onset symptom of all six patients in our group is abnormal gait caused by ankle dorsiflexion weakness, suggesting its diagnostic value. Meanwhile, weakness of finger extension is also very common. Axial muscles involvement is another feature of this disease, which is also seen in our patients that five showed weakness of neck flexion and two showed scoliosis [18, 19]. Other orthopedic abnormalities including Achilles tendon contracture, winging scapula, and talipes cavus have also been reported in previous researches [5, 20].
None of our patients showed evidence of cardiac involvement, but two presented with obvious sleep apnea, and one of whom even needed ventilation assistance during sleep. This symptom has not been reported in previous researches, but should be paid more attention. Therefore, we suggest regular polysomnography test in patients with Laing distal myopathy to detect early sleep apnea. Deep tendon reflexes were absent in two patients, in accordance with neurogenic lesions on their EMGs, which suggests possible lower motor neuron or peripheral nerve involvement. High amplitude motor unit potentials and spontaneous activities on EMGs in Laing distal myopathy have also been pointed out in several reports, but without definite explanations [16, 17]. Muscle biopsy in patient III-1 (family C) showing fiber type grouping further supports the evidence of denervation.
Muscle MRI in our patients shows both similarities and distinctions. In the thigh level, there is a tendency of concentric distribution of fatty infiltration in anterior muscles with vastus intermedius more severe than other quadriceps. Adductor magnus is affected the most among thigh adductor muscles, but posterior thigh muscles are affected differently among patients. This distribution is similar to previous researches that vastus intermedius and lateralis were reported to be the most affected muscles followed by biceps femoris and semimembranosus, whereas the rectus femoris, adductor longus, semitendinosus, and gracilis were usually spared [8, 21]. In the calf level, tibialis anterior and extensor hallucis longus are the most severely affected, which is in accordance with clinical features of ankle dorsiflexion weakness and also the typical “hanging big toe” sign [9, 22]. The early involvement of tibialis anterior has also been reported in several researches, but our study further emphasizes extensor hallucis longus is also affected at the early stage [8]. Besides, one of our patients also presented with fatty infiltration of axial muscles, which was also seen in other studies [19]. Meanwhile, the asymmetrical involvement of both limbs is present in our patients, which is also seen in previous reports, and this could be another MRI feature of the disease [19].
Muscle pathological changes in our patients including type 2 fiber predominance, multi-cores and abnormal mitochondria, were relatively moderate and non-specific, which were in accordance with previous researches [8, 9, 17, 23]. Multi-cores were seen on muscle biopsy of patient II-2 (family D), and were also reported in some previous articles in patients with the same MYH7 mutation p.E1508del [16, 24, 25]. This could be a specific pathological feature of this genotype. Other reported pathological features of the disease include central core, rimmed vacuoles et al. This suggests that muscle pathology could not be the definite diagnostic criteria of Laing distal myopathy but may only provide clues [26, 27]. Muscles selected for biopsy also affect the results that pathological changes of biceps were relatively slighter than tibialis in our study. Therefore, choosing more severely affected muscles for biopsy is also important, which can be assisted by MRI or other imaging techniques.
In four mutations of the MYH7 gene detected in our patients, three are in-frame deletions. Referring to published articles on Laing distal myopathy, common mutation types also include missense mutations, and in-frame indels. The p.K1617del and p.E1508del have also been reported in a few studies [5, 6, 8, 9, 12, 16, 20,21,22].Besides these in-frame indels in our study, other reported mutations include p.E1687del, p.K1729del, p.E1669del, p.K1729dup, p.L1793del, p.K1784del [5, 9, 11, 17, 27,28,29]. Therefore, in-frame deletions or duplications are common types of mutations in Laing distal myopathy. One presumable mechanism is that the mutations may change the microsatellite allele size [6, 30].
Conclusion
Our study further expands the clinical phenotypes and genotypes of Laing distal myopathy. In addition to muscle strengths and cardiac monitoring, we stress the importance of respiratory evaluation, especially polysomnography, in order to find early sleep apnea. Concentric fatty infiltration of anterior thigh muscles together with early involvement of tibialis anterior and extensor hallucis longus on MRI might be an imaging feature of the disease. Meanwhile, in-frame deletions of the MYH7 gene are common causes of Laing distal myopathy.
Availability of data and materials
The datasets used and/or analyzed during this study are available from the corresponding author upon request.
References
Guhathakurta P, Prochniewicz E, Thomas DD. Actin-myosin interaction: structure, function and drug discovery. Int J Mol Sci 2018, 19(9).
Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta. 2000;1496(1):3–22.
Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013;125(1):3–18.
Colegrave M, Peckham M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat Rec (Hoboken). 2014;297(9):1670–80.
Lamont PJ, Wallefeld W, Hilton-Jones D, Udd B, Argov Z, Barboi AC, Bonneman C, Boycott KM, Bushby K, Connolly AM, et al. Novel mutations widen the phenotypic spectrum of slow skeletal/beta-cardiac myosin (MYH7) distal myopathy. Hum Mutat. 2014;35(7):868–79.
Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, Duff RM, Beckman K, de Visser M, van der Graaff MM, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet. 2004;75(4):703–8.
Mastaglia FL, Phillips BA, Cala LA, Meredith C, Egli S, Akkari PA, Laing NG. Early onset chromosome 14-linked distal myopathy (Laing). Neuromuscul Disord. 2002;12(4):350–7.
Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F, Pedemonte M, Trovato R, Ruggiero L, Vercelli L, et al. MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis. 2016;11(1):91.
Lamont PJ, Udd B, Mastaglia FL, de Visser M, Hedera P, Voit T, Bridges LR, Fabian V, Rozemuller A, Laing NG. Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry. 2006;77(2):208–15.
Liu XY, Zhang YS, Sun AP, Zhong YF, Zheng DF, Fan DS. A novel MYH7 mutation resulting in Laing distal myopathy in a Chinese family. Chin Med J (Engl). 2019;132(7):856–9.
Li N, Zhao Z, Shen H, Bing Q, Guo X, Hu J. MYH7 mutation associated with two phenotypes of myopathy. Neurol Sci. 2018;39(2):333–9.
Oda T, Xiong H, Kobayashi K, Wang S, Satake W, Jiao H, Yang Y, Cha PC, Hayashi YK, Nishino I, et al. A de novo mutation of the MYH7 gene in a large Chinese family with autosomal dominant myopathy. Hum Genome Var. 2015;2:15022.
Xie Z, Xiao J, Zheng Y, Wang Z, Yuan Y. Magnetic resonance imaging findings in the muscle tissue of patients with Limb girdle muscular dystrophy type 2I harboring the founder mutation c.545A>G in the FKRP gene. Biomed Res Int. 2018, 2018:3710814.
Yu M, Zheng Y, Jin S, Gang Q, Wang Q, Yu P, Lv H, Zhang W, Yuan Y, Wang Z. Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS ONE. 2017;12(4):e0175343.
Waldmüller S, Erdmann J, Binner P, Gelbrich G, Pankuweit S, Geier C, Timmermann B, Haremza J, Perrot A, Scheer S, Wachter R, Schulze-Waltrup N, Dermintzoglou A, Schönberger J, Zeh W, Jurmann B, Brodherr T, Börgel J, Farr M, Milting H, Blankenfeldt W, Reinhardt R, Özcelik C, Osterziel KJ, Loeffler M, Maisch B, Regitz-Zagrosek V, Schunkert H, Scheffold T. German competence network heart failure: novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: results from the German Competence Network Heart Failure. Eur J Heart Fail. 2011;13(11):1185–92.
Dubourg O, Maisonobe T, Behin A, Suominen T, Raheem O, Penttila S, Parton M, Eymard B, Dahl A, Udd B. A novel MYH7 mutation occurring independently in French and Norwegian Laing distal myopathy families and de novo in one Finnish patient. J Neurol. 2011;258(6):1157–63.
Muelas N, Hackman P, Luque H, Garces-Sanchez M, Azorin I, Suominen T, Sevilla T, Mayordomo F, Gomez L, Marti P, et al. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology. 2010;75(8):732–41.
Dabaj I, Carlier RY, Gomez-Andres D, Neto OA, Bertini E, D’Amico A, Fattori F, PeReon Y, Castiglioni C, Rodillo E, et al. Clinical and imaging hallmarks of the MYH7-related myopathy with severe axial involvement. Muscle Nerve. 2018;58(2):224–34.
Park JM, Kim YJ, Yoo JH, Hong YB, Park JH, Koo H, Chung KW, Choi BO. A novel MYH7 mutation with prominent paraspinal and proximal muscle involvement. Neuromuscul Disord. 2013;23(7):580–6.
Van den Bergh PY, Martin JJ, Lecouvet F, Udd B, Schmedding E. Laing early-onset distal myopathy in a Belgian family. Acta Neurol Belg. 2014;114(4):253–6.
Pegoraro E, Gavassini BF, Borsato C, Melacini P, Vianello A, Stramare R, Cenacchi G, Angelini C. MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromuscul Disord. 2007;17(4):321–9.
Voit T, Kutz P, Leube B, Neuen-Jacob E, Schroder JM, Cavallotti D, Vaccario ML, Schaper J, Broich P, Cohn R, et al. Autosomal dominant distal myopathy: further evidence of a chromosome 14 locus. Neuromuscul Disord. 2001;11(1):11–9.
Cullup T, Lamont PJ, Cirak S, Damian MS, Wallefeld W, Gooding R, Tan SV, Sheehan J, Muntoni F, Abbs S, et al. Mutations in MYH7 cause Multi-minicore Disease (MmD) with variable cardiac involvement. Neuromuscul Disord. 2012;22(12):1096–104.
Reis GF, de la Motte G, Gooding R, Laing NG, Margeta M. Complex sarcolemmal invaginations mimicking myotendinous junctions in a case of Laing early-onset distal myopathy. Neuropathology. 2015;35(6):575–81.
Naddaf E, Waclawik AJ. Two families with MYH7 distal myopathy associated with cardiomyopathy and core formations. J Clin Neuromuscul Dis. 2015;16(3):164–9.
Romero NB, Xie T, Malfatti E, Schaeffer U, Bohm J, Wu B, Xu F, Boucebci S, Mathis S, Neau JP, et al. Autosomal dominant eccentric core disease caused by a heterozygous mutation in the MYH7 gene. J Neurol Neurosurg Psychiatry. 2014;85(10):1149–52.
Roda RH, Schindler AB, Blackstone C, Mammen AL, Corse AM, Lloyd TE. Laing distal myopathy pathologically resembling inclusion body myositis. Ann Clin Transl Neurol. 2014;1(12):1053–8.
Zima J, Eaton A, Pal E, Till A, Ito YA, Warman-Chardon J, Hartley T, Cagnone G, Melegh BI, Care4Rare C et al. Intrafamilial variability of limb-girdle muscular dystrophy, LGMD1D type. Eur J Med Genet 2020; 63(2):103655.
Muelas N, Hackman P, Luque H, Suominen T, Espinos C, Garces-Sanchez M, Sevilla T, Azorin I, Millan JM, Udd B, et al. Spanish MYH7 founder mutation of Italian ancestry causing a large cluster of Laing myopathy patients. Clin Genet. 2012;81(5):491–4.
Xu X, Peng M, Fang Z. The direction of microsatellite mutations is dependent upon allele length. Nat Genet. 2000;24(4):396–9.
Acknowledgements
The authors would like to acknowledge Ms. Yuehuan Zuo, Ms. Jing Liu and Ms. Qingqing Wang (Peking University First Hospital) for their works in preparations for pathological sections.
Funding
This work was supported by Scientific Research Seed Fund of Peking University First Hospital (2018SF032).
Author information
Authors and Affiliations
Contributions
MY and ZW conceived and planned this study and take full responsibility for the paper. MY, YL, HL, WZ, YY and ZW contributed to the analysis and interpretation of the clinical and genetic data. YZ reviewed the muscle MRI images, and contributed to the analysis and interpretation of the radiological data. MY took the lead in writing the manuscript. ZW supervised this study and contributed to revision of the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Human Research Ethics Committee of Peking University First Hospital, and all participants provided written informed consent.
Consent for publication
The patients described in this article provided consent for participation in the study and for publishing the obtained results.
Competing interests
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, M., Zhu, Y., Lu, Y. et al. Clinical features and genotypes of Laing distal myopathy in a group of Chinese patients, with in-frame deletions of MYH7 as common mutations. Orphanet J Rare Dis 15, 344 (2020). https://doi.org/10.1186/s13023-020-01626-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01626-y