
Abstract
Background
Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb. are ferns that are popularly used for medicine, as recorded by the Chinese pharmacopoeia, and are distributed in different regions of China. However, O. japonica is not record in the Standards of Chinese Herbal Medicines in Hong Kong. Research on identification methods of D. crassirhizoma and O. japonica is necessary and the phylogenetic position of the two species should be identified. The plastid genome is structurally highly conserved, providing valuable sources of genetic markers for phylogenetic analyses and development of molecule makers for identification.
Methods
The plastid genome DNA was extracted from both fern species and then sequenced on the Illumina Hiseq 4000. Sequences were assembled into contigs by SOAPdenovo2.04, aligned to the reference genome using BLAST, and then manually corrected. Genome annotation was performed by the online DOGMA tool. General characteristics of the plastid genomes of the two species were analyzed and compared with closely related species. Additionally, phylogenetical trees were reconstructed by maximum likelihood methods. The content of dryocrassin of the two species were determined according to the Standards of Chinese Herbal Medicines in Hong Kong.
Results
The genome structures of D. crassirhizoma and O. japonica have different characteristics including the genome size, the size of each area, gene location, and types. Moreover, the (simple sequence repeats) SSRs of the plastid genomes were more similar to other species in the same genera. Compared with D. fragrans, D. crassirhizoma shows an inversion (approximately 1.6 kb), and O. japonica shows two inversions (1.9 kb and 216 bp). The nucleotide diversity (polymorphism information, Pi) analysis showed that the psbK gene and rpl14-rpl16 region have the highest Pi value in Dryopteris, and the ycf2-CDS3 and rpl14-rpl16 regions show the highest Pi vale in O. japonica. Phylogenetic analyses showed that the two species were grouped in two separate clades from each other, with both individually located with other members of their genus. The marker content of dryocrassin is not found in O. japonica.
Conclusions
The study is the first to identify plastid genome features of D. crassirhizoma and O. japonica. The results may provide a theoretical basis for the identification and the application of the two medically important fern species.
Similar content being viewed by others


Background
The chloroplast is the key organelle for photosynthesis and carbon fixation; chloroplasts also play vital roles in other aspects of plant physiology and development, including the synthesis of amino acids, nucleotides, fatty acids, phytohormones, vitamins, metabolites, and the assimilation of sulfur and nitrogen [1, 2]. In general, plastid genomes are structurally highly conserved across land plants, thus are valuable sources of genetic markers for phylogenetic analyses because of their very low level of recombination [3,4,5]. Some regions can also be used as DNA barcodes, such as psbH and rbcL, which provide the necessary universality and species discrimination [6]. Nguyen et al. developed many authentication markers for five major Panax species via comparative analysis of complete plastid genome sequences [7]. Aconitum coreanum (Levl.) Rapaics also contains a barcoding target sequence in a divergent region, ndhC–trnV, and a sequence characterized amplified region (SCAR) marker was successfully developed for its discrimination [8]. Comparative plastid genomic studies also provide an invaluable source of information for understanding plant evolution and phylogenetics [9]. Therefore, many studies on the plastid genome from different species have been reported. These studies have shown that the plastid genome gene content and orientation are considerably conserved between species [10]; however, there are also some changes such as in the size, gene intron gains and losses, expansion/contraction of inverted repeats (IRs), structure rearrangements, and inversions [11, 12]. A previous report suggested that the complete plastid genome sequence of ferns has undergone two major rearrangements, distinguishing higher leptosporangiate ferns from basal fern lineages [3]. The IRs of Polypodiales plastomes are dynamic, driven by such events as gene loss, duplication, and putative lateral transfer from mitochondria [13]. The fern also shows the major reduction in the rate of evolution, and there has been a major slowdown in the rate of mutation in Dicksonia squarrosa and Tmesipteris elongata [14]. Ferns also hold a critical phylogenetic position as the extant sister group to seed plants [15]. Therefore, understanding the organization and structure of fern plastid genomes provides useful information for the studies of ferns.
For Chinese herbal medicines plastid genes, such as psbA-trnH, rbcL, and matK, are usually used as DNA barcodes for plants [16]. In addition, trnL-F has also been used to study the phylogeography and demographic history of Chrysanthemum indicum L. [17]. However, DNA barcodes have limited resolution at the species level. The plastid genome of the common Chinese medicine Panax notoginseng (Burk.) F.H. Chen from different producing areas sometimes shows differences in length. Specifically, in Wenshan, Yunnan Province, the plastid genome size is 156,466 bp (Genbank Number: KJ566590) and in Wuliang Mountain, Yunnan Province, the plastid genome size is 156,324 bp (Genbank Number: KT001509). However, there is little structural difference between the two samples [18, 19]. This also occurs in C. indicum from difference regions (Genbank Number: JN867592, NC_020320).
Ferns are a large group of vascular plants with approximately 2129 species in China [20]. The family includes many medicinally important species such as Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb.. The roots of these species are widely used in Traditional Chinese Medicine with the effect of clearing heat, detoxifying, and deworming, and the effects are recorded in the Chinese pharmacopoeia [21]. D. crassirhizoma is distributed in northeast and northern China, and O. japonica is the most common species of ferns in warm temperate and subtropical regions of China [22, 23]; its young leaves are rich in nutrients and edible as a wild vegetable. The two species have the same effect described in the Chinese pharmacopoeia; however, there is a difference in the chemical composition of the two species. Dryocrassine is the index component of D. crassirhizoma [24], and osmundacetone is the specialty component of O. japonica [25]. The species are also used in different prescriptions. Specifically, O. japonica is not record in the Standards of Chinese Herbal Medicines in Hong Kong [24]. Previous studies have reported the identification of D. crassirhizomatis based on plastid barcodes using psbA-trnH. This barcode can identify D. crassirhizomatis and its adulterants [26]. In addition, psbA-trnH and rbcL can distinguish O. japonica from its adulterants [27]. The O. japonica plastid genome regions, including rbcL, accD genes for ribulose 1,5-bisphosphate carboxylase/oxygenase, acetyl-CoA carboxylase beta subunit, and partial cds, have been reported (GenBank: AB494712.1). Even though studies on the plastid genes of these two species have been reported, their phylogenetic positions are vague and the identification from closely related species based on barcodes has not been reported. A comparative analysis about the content of dryocrassin in the two species is unclear. Here, we determined the dryocrassin components of the two species and sequenced and analyzed the complete plastid genomes of D. crassirhizomatis (Dryopteridaceae) and O. japonica (Osmundaceae). A comparative analysis was conducted with the closely related species to provide information for the quality control of related medicinals and allow for a better understanding of plastid genome evolution within their respective families.
Methods
DNA extraction and sequencing
We collected leaves from one individual of D. crassirhizoma and O. japonica, respectively, from Qianshan, Anshan City, Liaoning Province (N40°39′47.42″, E124°52′13.16″) and Sanming, Jianning City, Fujian Province (N27°49′8.06″, E117°43′10.93″) China. The two species were identified by Kang Tingguo, a professor from Liaoning University of Traditional Chinese Medicine. Voucher specimens were deposited in the Liaoning University of Traditional Chinese Medicine Herbarium (D. crassirhizoma 20170827001LY, O. japonica 20170917001LY).
Plastid DNA was extracted from approximately 5 g fresh, young leaves from D. crassirhizoma and O. japonica using a modified cetyl trimethylammonium bromide method [28]. After DNA isolation, 1 μg of purified DNA was fragmented and used to construct short-insert libraries (insert size 430 bp) according to the manufacturer’s instructions (Illumina), then sequenced on an Illumina Hiseq 4000 [29]. Prior to assembly, raw reads were filtered. This filtering step was performed to remove reads with adaptors, reads showing a quality score below 20 (Q < 20), reads containing a percentage of uncalled based (“N” characters) equal or greater than 10%, and the duplicated sequences. The plastid genome was reconstructed using a combination of de novo and reference-guided assemblies, and the following three steps were used to assemble plastid genomes [30]. First, the filtered reads were assembled into contigs using SOAPdenovo2.04 [31]. Second, contigs were aligned to the reference genome of two species using BLAST, and aligned contigs (≥ 80% similarity and query coverage) were ordered according to the reference genome. Third, clean reads were mapped to the assembled draft plastid genome to correct any wrong bases, and the majority of gaps were filled through local assembly.
Genome assembly and annotation
The plastid genes were annotated using the online DOGMA tool (http://dogma.ccbb.utexas.edu/index.html) [32], using default parameters to predict protein-coding genes, transfer RNA (tRNA) genes, and ribosome RNA (rRNA) genes. A whole plastid genome BLAST [33] search (E-value ≤ 1e−5, minimal alignment length percentage ≥ 40%) was performed against five databases: KEGG (Kyoto Encyclopedia of Genes and Genomes) [34,35,36], COG (Clusters of Orthologous Groups) [37, 38], NR (Non-Redundant Protein Database databases), Swiss-Prot [39], and GO (Gene Ontology) [40]. Sequencing data and gene annotation were then submitted to GenBank and assigned accession number (D. crassirhizoma: MK554795, O. japonica: MK554796).
Plastid genome mapping
The plastid genomes of D. crassirhizoma and O. japonica were exported in GenBank format and the plastid genome was mapped using Organellar Genome Draw (OGDRAW) (Max Planck Institute of Molecular Plant Physiology, Am Mühlenberg, Potsdam, Germany) (http://ogdraw.mpimp-golm. mpg.de/index. Shtml) [41].
Comparative analysis of genomes
The simple sequence repeat (SSR) software MicroSAtellite (MISA) (http://pgrc.ipk-gatersleben.de/misa/) was used to identify the SSR sequences, and tandem repeats of 1–6 nucleotides were considered microsatellites. The minimum number of repeats were set to 8, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. The data was compared with Osmundastrum cinnamomeum L. (O. cinnamomea L.) (NC_024157.1), D. fragrans (L.) Schott (KX418656), and D. decipiens (Hook.) O. Ktze. (NC_035854.1). We focused on perfect repeat sequences [42]. Long repeat sequences of the two species were identified using REPuter. Four types of repeats (dispersed, tandem, palindromic, and gene similarity repeats) were determined. The maximal length of the gap size between palindromic repeats was 3 kb. Overlapping repeats were incorporated into one repeat motif whenever possible and a given region in the genome was defined as having only one type of repeat. When one repeat motif could be described as both tandem and dispersed, the region was described as a tandem repeat rather than a dispersed repeat [43]. The alignments were visually checked and edited manually. The gene order of the plastid genomes of D. crassirhizoma and O. japonica were compared with D. fragrans using online zPicture software (http://zpicture.dcode.org/) [44]. To screen variable characters between D. crassirhizoma, D. fragrans, and D. decipiens and between O. japonica and O. cinnamomeum, the average number of nucleotide differences and total number of mutations were determined to analyze nucleotide diversity (polymorphism information, Pi) using DnaSP v5.0 [45].
Phylogenetic analysis
To identify the phylogenetic position of D. crassirhizoma and O. japonica and their relationship with other families, phylogenetic trees were constructed by the plastid genome sequences from 30 species, and 28 species were download from GenBank. Among them, two species, Selaginella uncinata (Desv.) Spring and Selaginella moellendorffii Hieron., were set as outgroups. The analysis was run using the whole plastid genomes single nucleotide polymorphisms (SNPs). A Maximum Likelihood (ML) phylogenetic tree was constructed using PhyML 3.0, and the model GTR + I + G was selected for the ML analyses with 100 bootstrap replicates to calculate the bootstrap values [46].
Comparative analysis of dryocrassin content in D. crassirhizoma and O. japonica
This research used high-performance liquid chromatographic detection the dryocrassin content in D. crassirhizoma and O. japonica [24]. The Additional file 1: Minimum Standards of Reporting Checklist includes details of the experimental design, statistics, and resources used in this study.
Results
Plastid genome structure
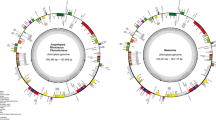
In the fully assembled plastid genome genome sequence from D. crassirhizoma and O. japonica, the genome size was 153,559 bp and 143,220 bp, the large single copy region (LSC) was 82,495 bp and 100,464 bp, the small single copy region (SSC) was 21,599 bp and 22,224 bp, the IRs were 49,464 bp and 20,532 bp, respectively (Table 1). The plastid genome structure of the two species showed differences in size of the same region (Fig. 1). There were 86 and 84 protein coding genes annotated for D. crassirhizoma and O. japonica, respectively. There were significant differences in length and genes of the IR region between the genome sequences (Fig. 1).

Plastid genome map for two sequenced ferns. Boxes on the inside (I) and outside of the outer circle (II) represent genes of D. crassirhizoma and O. japonica, respectively
D. crassirhizoma contains 31 tRNA genes, 10 rRNA (Additional file 2: Table S1), and 12 tRNA genes, and all rRNAs were located in the IR region (Additional file 2: Table S1). O. japonica contains 36 tRNA genes, eight rRNA genes, and 14 tRNAs in the IR region. The number of tRNAs in the two species were different in that D. crassirhizoma had five fewer tRNAs than O. japonica. The gene trnS-CGA had two copies in O. japonica and was in the IR region, but in D. crassirhizoma, only one copy was present. The gene trnH-GUG showed an opposite phenomenon. The genes trnK-UUU, trnL-CAA, trnR-CCG, trnT-GGU, and trnV-GAC were not present in D. crassirhizoma and the genes trnT-GGU and trnV-GAC were located in the IR region. The genes trnT-UGU and trnL-UAA were not present in O. japonica. The protein coding gene ndhB, psaM, and rps14 were only annotated in O. japonica but the gene ycf1 was absent. The genes ycf2, rps7, psbA, rps12, and psbA were located in the IR region in D. crassirhizoma.
Among the protein coding genes, D. crassirhizoma was found to have 20 genes contain the introns while O. japonica have 12 (Table 2). In D. crassirhizoma, the gene ycf2 encoded four introns, the gene rpoB encoded three introns, and the genes clpP and ndhF encoded two introns. In O. japonica the genes clpP, ycf2, and ycf3 encoded two introns.
Repeat sequences analysis
SSR sequence analysis
A total of 74 and 82 SSR locis that were 1024 bp and 1191 bp long, respectively, were detected in the D. crassirhizoma and O. japonica plastid genomes (Table 3). The number of mono-repeats were dominant in the plastid genomes of both species. There were 54 and 62 SSRs located in the LSC, 14 and 12 located in the IR, and six and eight located in the SSC in the D. crassirhizoma and O. japonica plastid genome, respectively (Additional file 3: Table S2, Additional file 4: Table S3). Compared with D. fragrans, D. decipiens, and O. cinnamomeum (Fig. 2), we found that the three species from Dryopteris (Dryopteridaceae) had more SSR mono-repeats than the two species from Osmunda.

SSRs in the plastid genomes of O. japonica, D. crassirhizoma, and three other fern species. Mono represents mononucleotide repeats, Di represents dinucleotide repeats, and Tri represents trinucleotide repeats
Large repeat analysis
Large repeat sequences showed repeats with lengths of ≥ 30 bp each. Twenty-six and 111 pairs of large repeat sequences with sequence identity of > 90% were found in the O. japonica (Additional file 5: Table S4) and D. crassirhizoma (Additional file 6: Table S5) plastid genomes. The repeats from D. crassirhizoma ranged from 30 to 145 bp in length, and in O. japonica, the repeats ranged from 30 to 46 bp in length. A total of 10 and 87 large repeat sequences were located in the genes in O. japonica and D. crassirhizoma, respectively.
Sequence order
Compared with D. fragrans, the D. crassirhizoma plastid genome showed a high degree of collinearity and translocation. Furthermore, it also showed low degree of transinversion (Fig. 3a). The D. crassirhizoma plastid genome shares one approximately 1.6 kb long inversion with the D. fragrans plastid genome. The inversion is located at the beginning of the IR between the trnS-CGA and ycf12. Compared to the D. fragrans plastid genome, the O. japonica plastid genome showed a high degree of collinearity (Fig. 3b). It also showed a low degree of transinversions. Compared with D. fragrans, O. japonica showed two inversions, with one approximately 1.9 kb long at the beginning of LSC and the other about 216 bp long located in SSC.

Comparisons of the gene order of the D. crassirhizoma (a) and O. japonica (b) plastid genome with the gene order of the plastid genome of D. fragrans
LSC, SSC, and IR border regions analysis
We analyzed the border structures of three Dryopteris species (Fig. 4a) and two Osmunda species (Fig. 4b). The lengths of the IR regions of the Dryopteris species were different and ranged from 17,321 to 24,732 bp, and the expansion and contraction of IR regions differed. There were 2673 bp, 72 bp, and 2673 bp no-coding regions in D. crassirhizoma, D. fragrans, and D. decipiens in the IRa/SSC boundary, respectively. The ndhF gene of D. fragrans was located in the LSC regions; the ndhF gene of the other two species extended into the IRa regions. The chlL gene was located in the SSC and extended into the IRb in D. crassirhizoma and D. decipiens. However, in D. fragrans there was no gene here. The gene ndhB located in the LSC and extended into IRb in D. fragrans. In D. crassirhizoma and D. decipiens there was no gene at the IRb/LSC border. The adjacent genes were matK.

Comparisons of LSC, SSC, and IR border regions among three Dryopteris species (a) and two Osmunda (b) species
The boundary characteristics of O. japonica and O. cinnamomeum were similar. The lengths of the IR regions of O. japonica and O. cinnamomeum were 10,265 bp and 10,109 bp. The ndhF genes of O. japonica located in the SSC region and extended into the IRa region and were 118 bp long. In O. cinnamomeum, there was a no-coding region (2110 bp) adjacent the IRa/SSC border.
Nucleotide diversity analysis
Plastid genome sequences contain regions that are highly variable. Such regions (coding and non-coding regions) are useful for the screening of suitable loci to resolve closely related species or genera in phylogenetic analyses and for DNA barcoding. The coding genes and non-coding regions of three Dryopteris species and two Osmunda species were compared (D. crassirhizoma was compared with D. fragrans and D. decipiens, and O. japonica was compared with O. cinnamomeum). We generated 107 coding genes (Fig. 5a) and 91 non-coding genes (Fig. 5b) within Dryopteris. Among the Pi values obtained from the comparative analysis (Additional file 7: Table S6), we found Pi values were ranged from 0.0000 (petG gene) to 0.20222 (psbK gene) of the coding genes, and most Pi values of the gene were greater than 0.01. The Pi value of non-coding genes loci ranged from 0.01380 (rps12-rps7-D2 region) to 0.13333 (rpl14-rpl16 region) of the non-coding genes loci. The coding gene regions were much more conserved than the non-coding gene regions. The first two significant variable loci were the gene psbK (Pi = 0.20222) and the rpl14-rpl16 region (Pi = 0.13333), and they both were located in the LSC region.

Comparative analysis of the nucleotide variability by Pi values within Dryopteris (a coding region, b non-coding region) and within Osmunda (c coding region, d non-coding region)
Within Osmunda, we generated 94 coding genes (Fig. 5c) and 101 non-coding genes (Fig. 5d). The Pi values ranged from 0.0000 (clpP-CDS3 gene) to 0.09942 (ycf2-CDS3 gene) of the coding genes and most Pi values of the genes were greater than 0.01 (Additional file 8: Table S7). Pi values were ranged from 0.01380 (rps12-rps7-D2 region) to 0.13333 (rpl14-rpl16 region) of the non-coding genes. The significant variable loci (ycf2-CDS3 gene and rps12-rps7-D2 region) were located in the LSC region.
Phylogenetic analysis
In this study, we investigated the phylogenetic relatedness among the plastid genomes of 30 species. There were 22 nodes with support values of 100% and four nodes with support values greater than 90%. The ferns of Leptosporangiatidae, Psilophytinae, and Equisetinae were grouped into three separate clades, respectively. The two Eusporangiate ferns were not grouped in one clade, with Mankyua chejuensis B.Y. Sun was closer to Psilophytinae and the other one Angiopteris evecta (G. Forst.) Hoffm. (Marattiaceae) was identified as a sister genus to Leptosporangiatidae. The two ferns we study were showed 100% homology with ferns from their respective families in the phylogenetic trees. The Leptosporangiatidae ferns formed two clades: Osmuda and the other clade contained the other species. This tree also indicated that the moss Physcomitrella patens (Hedw.) Bruch & Schimp was grouped in one clade with Leptosporangiatidae, Psilophytinae, Equisetinae, and Eusporangiate (Fig. 6).

Phylogenetic relationships of 30 species. The ML method used 1000 reiterations for the bootstrapping analysis
Comparative analysis of dryocrassin content in D. crassirhizoma and O. japonica
We were able to determine the content dryocrassin in D. crassirhizoma. The content has the same relative retention time as the marker content dryocrassin when detected by HPLC, but dryocrassin was not detected from the O. japonica.
Discussion
Plastid genome structure
Searching the whole plastid genome to identify the most variable regions and focusing on regions for mini-barcodes are believed to be efficient methods for developing taxon-specific DNA mini-barcodes [18]. And the identification of a medicinal material at the DNA level provides an objective and powerful tool for quality control [47]. For Chinese herbal medicine, there is insufficient evidence if Chinese herbal medicines from different producing areas would show differences in the plastid genome. The plastid genome of the same species from different regions does not show significant differences, but we could see that variation still exists [18, 19]. And the universal conventional DNA barcodes developed from the plastid genomes can rarely be used for identification within species. But, for many plant species, comparing the plastid genome with related species shows relatively conservative gene content [48] and also can be identified by the universal conventional DNA barcodes developed from the plastid genomes [7, 8]. We thought studying the plastid genome may significant in studying the origin traceability of Traditional Chinese Medicine.
The total size of the D. crassirhizoma plastid genome was slightly little difference from the plastid genome of another species within the same genus (D. fragrans, KX418656, 151,978 bp and D. decipiens, NC_035854.1, 150,987 bp) [49]. This is similar to Amomum compactum Soland ex Maton, which has a complete plastid genome is different from slightly from the other close relative species [50]. The total size of the O. japonica genome was larger than the genome size of Osmunda species (O. cinnamomeum, NC_024157.1, 142,812 bp) [9]. Such differences in plastid genome size may result from the expansion and contraction of the border areas between IR regions and single copy regions [51]. The related ferns had a relatively conservative genome size that was similar to many Angiosperms [8, 48].
Compared to O. japonica the tRNA genes trnK-UUU, trnL-CAA, trnR-CCG, trnT-GGU, and trnV-GAC were not present in D. crassirhizoma. All of these tRNA genes, except trnT-GGU, were also not present in the Cyrtomium falcatum (L.f.) Presl [51]. However, trnI-GAU was present in D. crassirhizoma and C. falcatum, but it was not present in many other Dryopteris species [13]. The trnR-CCG was present in O. japonica, which was consistent with previous studies that detected intact one in Gleicheniales, Hymenophyllales, and Osmundales [3, 52]. The genes trnT-UGU and trnL-UAA were not present in O. japonica. The trnT-UGU was also detected in Alsophila spinulosa (Wall. ex Hook.) R. M. Tryon [3]. The protein gene psaM was present in O. japonica but not in D. crassirhizoma. This gene also been detected in Psilotum [53] and Angiopteris [54], but not in Adiantum [55], suggesting that variation in genes is common.
The introns in eukaryotes can be applied to study phylogenetic evolution, evolutionary distance, and the regulation of gene expression [56]. Among the protein coding gene genes, D. crassirhizoma had more genes contained introns. The two ferns both had the RpoC1 intron, but many ferns of the genus Lygodium have lost the RpoC1 intron [9]. Those plastid genome modifications, such as gene/intron gains or losses, can be used to describe characters.
Repeat sequences analysis
Plastid genome SSRs are also the effective molecular markers to study polymorphisms and have been used within sunflower (Helianthus annuus) [57]. The results show that D. crassirhizoma had more SSRs in its plastid genome than O. japonica. The three Dryopteris species had more mono-repeat SSRs than the plastid genomes of the two species from Osmunda. Within the same family, the species have similar SSR compositions. This may also demonstrate the conservativeness of the plastid genome [3]. We also found numerous of long repeated sequences in D. crassirhizoma and O. japonica. The lengths of the repeats found in O. japonica represent much shorter repeats than those in D. crassirhizoma. The majority of long repeated sequences were located in noncoding regions that have been reported in several Angiosperm lineages [58]. Our data will contribute to further research on population genetics and phylogeography of these two fern genera.
Sequence order
Rearrangements of sequence order in fern have been reported before [3]. The two ferns were compared with D. fragrans. They all showed a high degree of collinearity and one or two inversions. The Alsophila plastid genome also has been reported shares three key inversions with other ferns relative to bryophytes [3]. All the inversions were located at the beginning of the IR, which may because of high variability in the IR sequence and gene content [14]. Inversions may very common within the ferns. This would contribute to the study of kinship of different species.
LSC, SSC, and IR border regions analysis
Although the IR regions are highly conserved, the expansion and contraction of IR regions is the general feature of plastid genomes, and they are mainly responsible for variations in plastid genome size and rearrangement [59, 60]. Dryopteris species have relatively different boundary characteristics with the length and the expansion and contraction of IR regions. This has mainly been reflected in the presence or absence of boundary genes and their size. The boundary characteristics of O. japonica and O. cinnamomeum were relatively similar; however, the two species we studied show great differences. The plastid genomes in closely related species also exhibit considerable variation in ferns, which was different from that in the Angiosperms [8]. However, these hypotheses need require the testing of more plastid genome sequences in the future.
Nucleotide diversity analysis
In the plastid, genome mutation events are usually gathered in “hot spots” and these mutational dynamics created highly variable regions dispersed throughout the plastid genome [61, 62]. The highly variable regions, trnH-psbA, trnR-atpA, atpI-rps2, rps2-rpoC2, petN-psbM, rps4-trnT, and rpl33-rps18, between Oresitrophe and Mukdeni have been report before [63]. Within Dryopteris the IR region was much more conserved than the LSC and SSC regions in both coding and non-coding regions, which was similar to previous studies [63]. Analysis of the five species indicated that the coding region was more conserved than the non-coding region. Non-coding sequences of the plastid genome are a primary source of data for molecular systematics, phylogeographic, and population genetic studies of plants [61], thus this would provide important genetic information for subsequent studies on phylogeography and divergence history of Dryopteris and Osmunda species.
Phylogenetic analysis
Plastid genome data are beneficial in resolving species definitions because organelle-based “barcodes” can be established for a species and then used to unmask interspecies phylogenetic relationships [58]. The phylogenetic relationships among many ferns have been studied through different methods, and at the broadest level, our results were congruent with previous studies [64, 65]. The ferns of Psilophytinae formed a sister clade to Equisetinae with strong support, which was different from a previous study [29]. It is thus necessary to expand taxon sampling as the next step in future phylogenomic analyses of polypods to confirm the position of ferns. The two ferns of eusporangiate were not grouped in one clade and M. chejuensis was more closely related to Psilophytinae, which was similar to previous reports [14]. The phylogenetic relationships among the two eusporangiate ferns are still uncertain [9]. The position of Osmuda may indicate that Osmuda diverged early in the lineage of leptosporangiate ferns [9]. The position of the two fern species that we study were consistent with morphological classification, thus the plastid genome may become an important assistant method for species classification.
Comparative analysis of dryocrassin content in D. crassirhizoma and O. japonica
We can determine the marker content of dryocrassin from D. crassirhizoma, but it was not detected from O. japonica. Even though they were both recorded in the Chinese Pharmacopoeia to have a similar curative effect [17], their chemical composition is different. It seems that they have different active sites, so the identification is necessary.
Conclusions
In this study, we conducted plastid genome skimming for D. crassirhizoma and O. japonica. By comparing and analyzing these data, we found the structure of the two plastid genomes was very different and the main active components are also different. Different SSR features may be able to be used to develop molecular markers for molecular identification and genetic diversity. The genomes also show a certain number of inversions and translocations when compared with the other fern species. In addition, the nucleotide diversity provides a reference for studying the genetic variation of the two species. The genomic structure and genetic resources presented in this study contribute to further studies on population genetics, phylogenetics, and conservation biology of ferns.
Abbreviations
- LSC:
-
large single copy
- SSC:
-
small single copy
- IR:
-
inverted repeat
- tRNA:
-
transfer RNA
- rRNA:
-
ribosomal RNA
References
Neuhaus HE, Emes MJ. Nonphoto synthetic metabolism in plastids. Annu Rev Plant Physiol Plant Mol Biol. 2000;51:111–40.
Daniell H, Lin CS, Yu M, Chang WJ. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016;17:134.
Gao L, Yi X, Yang Y, Su Y, Wang T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern cp genomes. BMC Evol Biol. 2009;9:130.
Provan J, Powell W, Hollingsworth PM. Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol Evol. 2001;16:142–7.
Ravi V, Khurana JP, Tyagi AK, Khurana P. An update on chloroplast genome. Plant Syst Evol. 2008;271:101–22.
Kress WJ, Erickson DL. A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS ONE. 2007;2:e508.
Nguyen VB, Park HS, Lee SC, Lee J, Park JY, Yang TJ. Authentication markers for five major Panax species developed via comparative analysis of complete chloroplast genome sequences. J Agric Food Chem. 2017;65:6298–306.
Park I, Kim W, Yang S, Yeo S, Li H, Moon BC. The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS ONE. 2017;12:e0184257.
Kim HT, Chung MG, Kim KJ. Chloroplast genome evolution in early diverged Leptosporangiate ferns. Mol Cells. 2014;37:372–82.
Choi KS, Park S. The complete chloroplast genome sequence of Aster spathulifolius (Asteraceae); genomic features and relationship with Asteraceae. Gene. 2015;572:214–21.
Liu H, Qian J, Li X, Sun Z, Xu X, Chen S. Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules. 2017;22:249.
Kim K, Choi K, Jansen RK. Two cp DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae). Mol Biol Evol. 2005;22:1783–92.
Logacheva MD, Krinitsina AA, Belenikin MS, Khafizov K, Konorov EA, Kuptsov SV, et al. Comparative analysis of inverted repeats of polypod fern (Polypodiales) plastomes reveals two hypervariable regions. BMC Plant Bio. 2017;17:255.
Zhong BJ, Fong R, Collins LJ, McLenachan PA, Penny D. Two new fern cp and decelerated evolution linked to the long generation time in tree ferns. Genome Biol Evol. 2014;6:1166–73.
Pryer KM, Schneider H, Smith AR, Cranfill R, Wolf PG, Hunt JS, Sipes SD. Horsetails and ferns are a monophyletic group and the closest living relatives to seed plants. Nature. 2001;409:618–22.
Chen YL, Fan G, Liu Y, Song C, Zhang Y, Xiang L, et al. Identification of traditional chinese medicine Rubiae radix et and its adulterants using DNA barcodes. Chinese Pharmaceutical Journal. 2015;50:1266–72.
Fang H, Guo Q, Shen H, Shao Q. Phylogeography of Chrysanthemum indicum L. (Compositae) in China based on trnL-F sequences. Biochem System Ecol. 2010;38:1204–11.
Dong W, Liu H, Xu C, Zuo Y, Chen Z, Zhou S. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 2014;15:138.
Zhang D, Li W, Gao C, Liu Y, Gao L. The complete plastid genome sequence of Panax notoginseng, a famous traditional Chinese medicinal plant of the family Araliaceae. Mitochondrial DNA. 2015;27:3438–9.
Wu Z, Raven PH, Hong D. Introduction. In Flora of China, Flora of China editorial board. Beijing: Science Press, St. Louis: Missouri Botanical Garden Press; 2013. p. 21.
Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia. 342nd ed. Beijing: China Medical Science and Technology Press; 2015. p. 300–31.
Wu S, Xiang J, Lu S, Wang F, Xing F, Dong S, etal. Dryopteridaceae. In Flora of China, Flora of China editorial board. Beijing: Science Press, St. Louis: Missouri Botanical Garden Press; 2013. p. 586.
Zhang X, Kunio I, Yoko K. Osmundaceae. In Flora of China, Flora of China editorial board. Beijing: Science Press, St. Louis: Missouri Botanical Garden Press; 2013. p. 588.
Department of health Hong kong Special Administrative Region People’s Republic of China. Dryopteridis Crassirhizomatis Rhizoma. In Hong Kong Chinese Materia Medica Standards. Hong kong: Department of health Hong Kong Special Administrative Region People’s Republic of China; 2013. p. 370–381.
Zhang D, Li B, Yang L, Fu M, Fang J. Isolation and determination of osmundacetone in Osmundae Rhizoma. Chin Pharm J. 2010;45:1612–4.
Cai Z, Wu Y, Xu L, Zhao R, Wang B, Kang T. Identification of Dryopteridis Crassirhizomatis Rhizoma based on psbA-trnH barcode. China J Chin Mater Med. 2016;41:4183–7.
Zheng SH, Ren WG, Wang ZH, Huang LF. Use of cp DNA barcodes to identify Osmunda japonica Thunb. and its adulterants. Plant Syst Evol. 2015;301:1843–50.
Mcpherson H, Van der Merwe M, Delaney SK, Edwards MA, Henry HR, McIntosh E, et al. Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 2013;13:8.
Borgstrom E, Lundin S, Lundeberg J. Large scale library generation for high throughput sequencing. PLoS ONE. 2011;6:e19119.
Cronn R, Liston A, Parks M, Gernandt DS, Shen RK. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by- synthesis technology. Nucleic Acids Res. 2008;36:e122.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAP denovo2: an empirically improved memory-efficient short-read denovo assembler. Gigascience. 2012;1:18.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–5.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–80.
Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–7.
Kanehisa M. A database for post-genome analysis. Trends Genet. 1997;13:375.
Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–7.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41.
Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011:bar009.
Ashburner MM, Ball CAC, Blake J, Botstein D, Butler H, Cherry JMJ, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
Lohse M, Drechsel O, Bock R. Organellar genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2009;25:1451–2.
Mayer C, Leese F, Tollrian R. Genome-wide analysis of tandem repeats in Daphnia pulex—a comparative approach. BMC Genome. 2010;11:277.
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–42.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, et al. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–7.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Shaw PC, Wong KL, Chan AWK, Wong WC, But PPH. Patent applications for using DNA technologies to authenticate medicinal herbal material. Chin Med. 2009;4:21.
Wang M, Cui L, Feng K, Deng P, Du X, Wan F, et al. Comparative analysis of Asteraceae chloroplast genomes: structural organization, RNA editing and evolution. Plant Mol Biol Rep. 2015;33:1526–38.
Wei R, Yan Y, Harris AJ, Kang J, Shen H, Xiang Q, et al. Plastid phylogenomics resolve deep relationships among Eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol Evol. 2017;9:1646–57.
Wu M, Li Q, Xu J, Li X. Complete chloroplast genome of the medicinal plant Amomum compactum: gene organization, comparative analysis, and phylogenetic relationships within Zingiberales. Chin Med. 2018;13:10.
Raman G, Choi KS, Park S. Phylogenetic relationships of the fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo island based on cp genome sequencing. Genes. 2016;7:115.
Gao L, Wang B, Wang ZW, Zhou Y, Su YJ, Wang T. Plastome sequences of Lygodium Japonicum and Marsilea crenata reveal the genome organization transformation from basal ferns to core leptosporangiates. Genome Biol. 2013;5:1403–7.
Guo W, Zhu A, Fan W, Mower JP. Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 2017;213:391–403.
Roper JM, Kellon HS, Wolf PG, Karol KG, Mandoli DF, Everett KDE, et al. The complete plastid genome sequence of Angiopteris evecta (G. Forst.) Hoffm. (Marattiaceae). Am Fern J. 2007;97:95–106.
Wolf PG, Rowe CA, Sinclair RB, Hasebe M. Complete nucleotide sequence of the cp genome from a leptosporangiate fern Adiantum capillus-veneris L. DNA Res. 2003;10:59–65.
Cao J. Research progress in eukaryotic intron. Bioprocess. 2011;1:9–12.
Wills DM, Hester ML, Liu AJ, Burke M. Chloroplast SSR polymorphisms in the Compositae and the mode of organellar inheritance in Helianthus annuus. Theor Appl Genet. 2005;110:941–7.
Yang JB, Yang SX, Li HT, Yang J, Li DZ. Comparative chloroplast genomes of Camellia Species. PLoS ONE. 2013;8:e73053.
Yang M, Zhang X, Liu G, Yin Y, Chen K, Yun Q, et al. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS One. 2010;5:e12762.
Raubeson LA, Peery R, Chumley TW, Dziubek C, Fourcade HM, Boore JL, et al. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics. 2007;8:174.
Dong W, Liu J, Yu J, Wang L, Zhou S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE. 2012;7:e35071.
Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94:275–88.
Liu L, Wang Y, He P, Li P, Lee J, Soltis DE, et al. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics. 2018;19:235.
Grewe F, Guo W, Gubbels EA, Hansen AK, Mower JP. Complete plastid genomes from Ophioglossum californicum, Psilotum nudum, and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol Biol. 2013;13:8.
Des Marais DL, Smith AR, Britton DM, Pryer KM. Phylogenetic relationships and evolution of extant horsetails, Equisetum, based on cp DNA sequence data (rbcL and trnL-F). Int J Plant Sci. 2003;164:737–51.
Authors’ contributions
Conceptualization, TK and WW; Formal analysis, CL; Methodology, BW; Software, YX; Writing—original draft, LX; Writing—review and editing, LX. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during the course of this study are included in this document or obtained from the appropriate author(s) at reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This research was funded by National Natural Science Foundation of China (General Program, Grant Numbers 81874338, 81773852) and the Liaoning Province Education Department (Liaoning Higher School Outstanding Young Scholar Growth Plan, Grant Number LJQ2014101).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1.
Minimum standards of reporting checklist.
Additional file 2: Table S1.
List of genes obtained from the D. crassirhizoma Nakai and O. japonca Thunb. plastid genome sequences.
Additional file 3: Table S2.
The SSR characteristic in Dryopteris crassirhizoma Nakai.
Additional file 4: Table S3.
The SSR characteristic in Osmunda japonica Thunb..
Additional file 5: Table S4.
Long repeat sequences in Osmunda japonica Thunb. plastid genome.
Additional file 6: Table S5.
Long repeat sequences in Dryopteris crassirhizoma Nakai plastid genome.
Additional file 7: Table S6.
The Pi value of coding regions and no-coding regions of Dryopteris.
Additional file 8: Table S7.
The Pi value of coding region and no-coding region of Osmunda.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, L., Xing, Y., Wang, B. et al. Plastid genome and composition analysis of two medical ferns: Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb.. Chin Med 14, 9 (2019). https://doi.org/10.1186/s13020-019-0230-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13020-019-0230-4