
Abstract
Background
Combustion of biodiesels in place of fossil diesel (FD) has been proposed as a method of reducing transport-related toxic emissions in Europe. While biodiesel exhaust (BDE) contains fewer hydrocarbons, total particulates and carbon monoxide than FD exhaust (FDE), its high nitrogen oxide and ultrafine particle content may still promote pulmonary pathophysiologies.
Main body
Using a complement of in vitro and in vivo studies, this review documents progress in our understanding of pulmonary responses to BDE exposure. Focusing initially on hypothesis-driven, targeted analyses, the merits and limitations of comparing BDE-induced responses to those caused by FDE exposure are discussed within the contexts of policy making and exploration of toxicity mechanisms. The introduction and progression of omics-led workflows are also discussed, summarising the novel insights into mechanisms of BDE-induced toxicity that they have uncovered. Finally, options for the expansion of BDE-related omics screens are explored, focusing on the mechanistic relevance of metabolomic profiling and offering rationale for expansion beyond classical models of pulmonary exposure.
Conclusion
Together, these discussions suggest that molecular profiling methods have identified mechanistically informative, novel and fuel-specific signatures of pulmonary responses to biodiesel exhaust exposure that would have been difficult to detect using traditional, hypothesis driven approaches alone.
Similar content being viewed by others

Background
In 2017, UK road users travelled 325.5 billion miles [1]; a figure that is forecast to increase by 75 billion by 2040 [2] and follow a trend that is being predicted globally [3]. Fossil fuel combustion contributes significantly to traffic-related air pollution, releasing pollutant gases (including nitrogen oxides (NOx), sulphur dioxides and carbon monoxide (CO)) and particulates [4]. As these composition derived particles are of respirable size [5,6,7], exist ubiquitously in urban areas and have been shown to induce inflammatory pulmonary pathophysiologies [8,9,10,11], regulators welcome interventions that reduce their emission.
Biodiesel is a liquid fuel produced through transesterification of vegetable oils, animal fats or waste cooking oil. This base-catalysed reaction is driven by adding alcohol to the feedstock [12], converting the parent product to its less viscous alkyl-ester. When blended (up to 20% volume) with fossil diesels (FD), biodiesels can power modern compression-ignition engines efficiently without demanding modified transport or storage measures [13, 14].
Biodiesel has a higher oxygen content than FD, encouraging more complete fuel combustion [13]. Coupled with an absence of aromatic and sulphurous compounds, the resulting emissions can contain 90% fewer unburned hydrocarbons, 75–90% fewer polycyclic aromatic hydrocarbons (PAHs), 43% less CO and 55% less particulate matter (PM) than FD exhaust (FDE) [15, 16], depending on the engine and combustion conditions used to burn the fuel. As these pollutants associate significantly with the incidence of airway infection and respiratory symptoms, onset and exacerbation of inflammatory pulmonary diseases and rates of respiratory hospital admissions in heavily trafficked areas [17,18,19,20], biodiesel usage has been encouraged in Europe, with the 2009/28/EC directive striving to replace 10% of transport fuel with biodiesel by 2020 [21].
Despite some improvements in emission profiles, BDE generally contains more NOx and ultrafine particles (PM0.1) than FDE [22] with particle emissions increasing with the chain length and degree of double bond saturation in the feedstock fatty acids [23]. Given that PM0.1 have been shown to exhibit greater inflammatory potential and oxidant capacity than larger particles [24, 25] and that both PM0.1 and NOx have been associated with mortality from respiratory diseases [26, 27], it is imperative that the impacts that biodiesel exhaust (BDE) exposure have on pulmonary health are characterised and communicated to policymakers. Furthermore, BDE contains a number of molecules that have the potential to induce pulmonary toxicity that are not found in FDE (or are present at significantly lower levels). Esters such as those found within BD can produce toxic, mutagenic and carcinogenic carbonyls when burnt in the presence of O2, with concentrations of formaldehyde, acetaldehyde, acrolein, acetone, propionaldehyde and butyraldehyde increasing linearly as the BD component of blends increases [28]. Additionally, BD fuel esters themselves have been detected in BDE, including several species that are known to induce pulmonary irritation [29].
Two complementary approaches have been employed to investigate particulate toxicity; traditional hypothesis-driven methods and, more recently, agnostic approaches that employ various untargeted omics platforms to identify global patterns of response to generate novel insights and new hypotheses. Traditional approaches are of considerable value to epidemiologists because they associate pathophysiological endpoints (including airway responsiveness and respiratory symptoms) with underlying molecular events, i.e. they support causal inference. Furthermore, with BD being proposed as a replacement for FD, they enable investigation of endpoints that have previously been validated as markers of particle toxicity allowing comparative toxicological assessment. The techniques are however, limited in their ability to uncover novel pathways, making them less informative for mechanistic studies of complex aerosols with novel constituents.
In contrast, high-throughput omics techniques characterise the expression of 100s–1000s of molecules, or 10s of thousands of transcripts, simultaneously, enabling additional molecular events to be tested for association with exposures in an untargeted manner (Fig. 1). This approach has significantly advanced characterisations of FDE toxicity, demonstrating, for example, that lung cells undergo a hierarchical oxidative stress response following exposure [30] and that impairment of macrophage phagocytic functions may be influenced by differential expression of genes implicated in cytoskeletal rearrangements [31]. Still less commonplace than the cheaper traditional techniques, omics screens are gaining popularity as methods of characterising pulmonary responses to BDE exposure.

Major techniques and technologies employed during molecular profiling. Typical numbers of molecules detectable by each platform (during a full platform analysis) are displayed in brackets
The information that hypothesis-driven studies have provided on biodiesel-induced pulmonary toxicity has been reviewed comprehensively [32, 33]. Following from this work, our review focuses on recent transitions towards omics-based assessments of BDE-induced pulmonary toxicity. The work aims to evaluate the effectiveness of including molecular screens in characterisations of pulmonary toxicity mechanisms and to comment on the usefulness of these data in terms of understanding whether BD blends are a ‘healthy’ replacement for pure FD. To produce these evaluations, studies of biodiesel toxicity were identified within the PubMed database (http://www.ncbi.nlm.nih.gov/sites/entrez) using the key words ‘biodiesel AND lung OR pulmonary’ combined with ‘toxicity/ inflammation/ genotoxicity/ transcriptomics/ mass spectrometry or omics’. Reports from human cohort studies, animal exposures or in vitro modelling were included for review while studies performed in plants or with raw biofuels were excluded from the analysis.
Main body
Hypothesis-driven approaches to exhaust toxicity
The London smog episode in 1952 was one of the events that initiated research into the toxicity of ambient pollution, and since that time, characterisation of pulmonary responses to inhaled xenobiotics has advanced greatly [34]. Much of our knowledge of the adverse effects that exhaust emissions exert upon the lung is derived from human experimental chamber studies of FDE exposure [35, 36], or exposures of individuals to micro-environments with high traffic related emissions [37,38,39]. Historically, these studies have sought to measure validated markers of pulmonary responses in a targeted manner and are supported by increasing evidence from the epidemiological literature that respiratory mortality and morbidity is strongly related to components within the airshed that reflect emissions from diesel exhaust, such as elemental and black carbon [40,41,42].
Experimental chamber studies that have examined responses to the full diesel aerosol: particulates, volatile organic species and co-pollutant gases (NO2, NOx and CO) have shown that diesel exhaust (100–300 µg/m3 PM10) breathed over short durations of 1–2 h (in individuals performing intermittent exercise) can elicit the full pathway of neutrophilic inflammation in the lung [39], from upregulation of endothelial adhesion molecules [39], expression of pro-inflammatory cytokines [39], such as IL-6 and Gro-α, to established airway neutrophilia in both the airway tissues and respiratory tract lining fluids [39]. These responses occur in parallel with increased mast cell numbers in the bronchial mucosa and lymphocytosis [39, 43].
In these studies, largely conducted on young heathy volunteers, the causal role of the combustion derived nanoparticles in respiratory responses have been established experimentally using particle traps [44]. In addition, experiments comparing engines running under idling versus under load (standard city cycle), which impacts on the relative proportion of elemental carbon to organic carbon, have yielded broadly similar results, although some evidence of lower airway eosinophilia was noted followed exposure to diesel exhaust from the engine under load. Underpinning these inflammatory responses in the airways, activation of redox sensitive pathways has been reported, most clearly through the increased nuclear localisation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and activator protein 1 (AP-1) [45], but also through the upregulation of the epidermal growth factor receptor (EGRF) in the bronchial epithelium [46].
Less clear, until very recently, has been the mechanism by which diesel exhaust could lead to an exacerbation of asthma symptoms. Early studies, simply looking at the inflammatory response of mild and moderate asthmatic exposures to diesel failed to demonstrate clear evidence of augmented allergic inflammation [36], contrasting with the evidence of upper airway inflammation and pulmonary irritation (measured as a sustained reduction in forced expiratory volume) seen in asthmatics exposed to high levels of FDE as they walked along Oxford Street, London, UK [39]. Some mechanistic basis for increased respiratory symptoms in response to diesel particulates has recently been reported by experimental evidence demonstrating that the organic constituents associated with diesel particles can activate airway sensory afferents; a response that could be inhibited using a transient receptor potential ankyrin-1 antagonist, provision of antioxidants (N-acetyl cysteine) and inhibition of the aryl hydrocarbon receptor (AhR) [46]. Additionally, work by Carlsten et al. has demonstrated that diesel challenge of allergic individuals appears to sensitise the airways to subsequent allergen challenge [47, 48]. Thus, collectively over the last 20 years, the evidence base that FDE can impact adversely on both the healthy and allergic airway has increased and constitutes a substantial mechanistic underpinning of the epidemiological findings of adverse health effects associated with exposure to traffic-derived pollutants, especially FDE [49].
Biodiesel toxicity: the influence of fossil diesel studies
A decade ago, few groups had explored the health impacts of BDE exposure; either from an epidemiological or mechanistic viewpoint and unlike for FDE, human exposure studies had not been performed. However, histological changes such as increases in the lung: body ratios had been observed in female rats that had been exposed repeatedly to 0.5 mg particles /m3 of 100% soybean BDE via inhalation for thirteen weeks, along with dose-dependent increases in pulmonary macrophage counts and particle loading at lower and intermediate doses (0.04 and 0.2 mg/m3) [50]. These changes were consistent with the development of inflammatory pulmonary lesions and bronchiolar metaplasia in the alveoli where macrophages remained particle-laden for 28 days [50]. In terms of cytotoxicity, the soluble organic fraction (SOF) of rapeseed methyl ester (RME) exhaust was identified as more potent than its FD counterpart when applied to L929 mouse fibroblasts; while in the Ames test, the SOF of FDE-PM was more mutagenic to bacteria than BDE-derived particulate matter (BDE-PM) [32, 51]. Mutagenicity tests were also performed on rat hepatocytes, indicating that differences in the mutagenic potential of FDE and BDE were less pronounced in mammalian cells compared to bacteria; potentially reflecting differences in their metabolic capabilities [51]. Taken together it was proposed that studies of BDE toxicity should continue to draw from characterisations of FDE toxicity [52]. Indeed, comparisons between the toxicological potential of alternative and existing products are common-place in many fields, [53] and most biodiesel-themed studies have employed this approach.
While studies have continued to explore the genotoxicity of BDE, relative to FDE in vitro, a comprehensive evaluation of carcinogenicity of BDE is not currently possible because of a lack of human exposure studies and animal bioassays. In vitro investigations have provided mechanistic associations between components of FDE and the mutagenic and genotoxic effects of FDE exposure, but the classification of FD emissions as carcinogenic to humans (Group 1) by the International Agency for Research on Cancer was enabled by sufficient evidence being available from epidemiological studies and animal carcinogenicity assays [54].
Regardless of the type of diesel fuel, genotoxicity is mainly associated with the particulate fraction [32]. In an early study, comparison of organic extracts of a BDE with a FDE found that the former had approximately half the bacterial mutagenic activity of the latter, a reduction that correlated with decreased concentrations of PAHs and nitro-PAHs [55]. Some, but not all, combinations of fuel and engine type result in lower concentrations of particulate emission for BD or BD blends than for standard FD fuels, accompanied by lower bacterial mutagenicity [32]. Much of the mutagenic activity is associated with PAHs and with nitro-PAHs. Under oxidative or nitro-reductive conditions, organic extracts of FD and BD emissions generate DNA adducts in vitro, with properties compatible with those of adducts formed by PAHs and nitro-PAHs [56]. In one study, adduct formation by a BD blend was ~ 50% of that of conventional diesel [56], but in another, DNA adduct formation by extracts of particulates from rapeseed oil combustion emissions was comparable to that of conventional diesel emissions [57].
When generated from a diesel engine fitted with a catalyst, an RME BDE, but not a hydrogenated vegetable oil (HVO) BDE had lower particulates than a FDE and lower genotoxicity, measured by the comet assay, in RAW 264.7 macrophages [58]. This was accompanied by lower formation of reactive oxygen species (ROS). Not all studies have found lower activity than BDE than with FDE. A comparison between BDE and FDE from light-duty engines found similar ROS production and comet formation in A459 cells [59]. When organic extracts of particle emissions of different biodiesel blends were tested in human bronchial epithelial BEAS-2B cells, the frequency of micronucleus formation was similar for neat biodiesel, neat diesel and a 30:70 blend of the two [60]. In an in vivo study in rats, inhalation exposure resulted in induction of detectable levels of bulky DNA adducts in the lungs in the case of FDE, but not with BDE, but differences in levels of 8-oxodGuo in DNA by the two emissions sources were not observed [61].
Consistent with the notion that FDE stimulates inflammatory and oxidative stress responses in pulmonary cells [62], it was demonstrated that the SOF of soybean alkyl ester PM was a more potent inducer of IL-6 and IL-8 secretion than the equivalent extract of FDE-PM in BEAS-2B bronchial epithelial cells [63]. This observation was supported by data demonstrating that emissions produced from a 50% RME blend also induced greater cytotoxicity and IL-6 release than FD-derived counterparts in BEAS-2B cells, even in the presence of a DPF. Interestingly, the authors of this study demonstrated that their results were dependent on the data being normalised to distance driven during sample collection, indicating that rate of particle emission is an important metric to consider during dosing [64]. In THP-1 monocyte-derived macrophages, PM from B20 soy methyl ester (SME) induced stronger toxicity than FDE-PM, as determined by quantification of ROS 1-2 h post-exposure [29]. Similarly, particles produced from HVO stimulated a more potent dose-dependent increase in TNF-α and MIP-2 secretion than FDE-PM in RAW 264.7 cells, reaching statistical significance after exposure to 50 μg/ml doses rather than 150 μg/ml. While RME particles also induced dose-dependent increases in TNF- α and MIP-2 secretion, the response was significantly less than for FDE-PM or SME BD [58] indicating variable inflammatory potency for BD produced from different feedstocks.
Regarding the ranking of FD and BD potency, considerable inter-study discrepancies have been observed in vivo. In a murine model, Yanamala et al. observed that PM from pure fatty acid methyl ester (FAME) emissions induced cellular infiltration into the lung to a significantly greater extent than FDE-PM. This inflammatory response was characterised by neutrophilia 1-day post-exposure, followed by macrophage influx at the 7-day timepoint. In addition, reductions in pulmonary cell viability were observed at an earlier timepoint after BD exposure (1 day, compared with 7 days for FD) and markers of oxidative damage (4-hydroxynonenal protein carbonyls) and remained visible for a longer period (28 days) [65]. While Fukagawa et al. also reported that mice exposed to PM from a B20 SME blend had greater levels of protein carbonyls in their lungs than mice exposed to FDE-PM, they did not detect fuel-dependent differences in cellular infiltration with the airways, as assessed by bronchoalveolar lavage [29]. In further contrast, studies performed by the U.S Environmental Protection Agency (EPA) found FDE-PM exposures to be more toxic than BDE-PM exposures, with mice exposed to FDE-PM for 4 h exhibiting an 8-fold increase in pulmonary neutrophil influx and MIP-2 secretion than mice exposed to PM2.5 from B20 or B100 SME blends (at 500 μg/m3 doses only) [66]. Using a model of Wistar Kyoto rats, the EPA also demonstrated that FDE induced greater pulmonary injury (as determined by increased BALF GGT activity) than BDE (100% or 20% blend) following 24 h exposures and increased neutrophilia after dosing for 4 weeks [67]. As with their murine study [66], these effects were only visible at the highest tested dose (500 μg/m3) [67] so may not be especially representative of real-world exposures which are estimated to fall between 15 and 20 μg/m3 for urban roadside PM2..5 [68]. Despite this, it may be useful to note that FDE-PM but not PM from B20 or B100 SME blends increased proliferation of resting peribronchiolar lymph node cells and production of T helper cell cytokines in a murine model of allergic inflammation after 4 weeks of repeated exposure at 500 μg/m3 doses [39]. Such data demonstrate the possibility that BDE is less likely than FDE to induce allergic responses in individuals with allergic airway conditions.
Both in vitro and in vivo, these inter-study discrepancies have been attributed to differences in the fraction of exhaust tested. In human alveolar epithelial cells, apoptosis, viability and inflammatory responses have all been shown to be altered adversely by the presence of exhaust gases, [69] with semi-volatile extracts reducing viability to a greater extent than PM in the case of palm FAME [70]. The physicochemical properties of particles have also been considered, with particle surface area, transition metal content and mass concentration and diameter all being shown to contribute to inflammatory and cytotoxic potential in vitro [69] [71]. Such parameters may vary with choice of engine type, percentage blend of fuels [70], inclusion of emission modifying technologies [58, 64] and driving conditions [72]; factors that can introduce inter-study differences to both the BDE and FDE samples. Additionally, the choice of feedstocks may influence BDE-PM characteristics considerably; fuels derived from waste cooking oil for example, are chemically distinct to those produced from raw vegetable oils due to the array of thermolytic reactions that modify the feedstock during heating [73]. Furthermore, pollutant samples produced by splash blending of pure fuels to achieve B20 and B50 blends (as employed in the EPA studies) tend to exhibit linear increases in BD-specific chemical components as BD content increases while commercially available blends do not [33]. While these linear relationships may assist relation of toxicological endpoints to specific emission components, it is important to consider that splash-blended fuels can contain novel components [33], thus PM from splash blended B20 could cause different biological effects to PM from commercial B20.
Adding to variation between in vivo analyses, are possibilities of species-dependent threshold effects. Rodent models of particle exposure are susceptible to pulmonary overload (a progressive reduction in particle clearance caused by loss of motility in alveolar macrophages that results in chronic inflammation, fibrosis and tumorigenesis [74]) as well as minimal effective dose thresholds for particle-induced inflammatory responses [75]. Choice of exposure methods for rodent studies can impact on both the dose-rate (tissue-wide and compartmentally specific) and clearance or particles within the lung; both factors that can influence the applicability of threshold effects to individual models. As an example, intratracheal instillation of TiO2 particles (especially within the ultrafine fraction) induces significantly greater cellular infiltration of BALF than inhalation of the same particles, partly due to the total intended dose being administered all at once, rather than across an extended period [76]. Based on exposure-response curve data covering non-interventional exposures to PM doses as low as 2 μg/m3, and observations of low rates of tumour development in chronically particle-exposed individuals, it has been suggested that these thresholds are not relevant for humans [75, 77]. This raises additional questions as to whether data from rodent models that explore particle potency can be applied simplistically to human exposure scenarios or compared reliably with results from human cohort studies. Regardless of the components that drive these inter-study differences, their existence highlights the necessity to consider outcomes of BDE exposure as absolute values. While comparisons between BDE and FDE toxicity assist decision-making regarding the option of replacing fossil fuels with renewable alternatives, very few studies have concluded that BDE exposure has no adverse effects on pulmonary health or function. As such, absolute values for BDE toxicity metrics may prove vitally informative for policymakers, indicating the maximum levels of exposure that can be tolerated by humans. Additionally, greater attention should be paid to the metrics used to determine particle dosing. Across the field of emissions toxicology, convenience and desire for comparability with historical studies has encouraged the widespread use of particle mass for this purpose. Increasingly however, surface area has been argued to relate better to particle-induced responses, including influx of immune cells and cytokine secretion in mice [75] and pulmonary overload in rats [74]. Importantly, the heightened inflammatory potential reported with PM0.1 compared with larger particles of equivalent composition, has been shown to disappear when exposure doses are equalised for surface area, rather than mass [78]. Given that BDE contains considerably greater proportions of PM0.1 than FDE, it may be prudent to consider surface area as a dose metric for future comparisons of toxicity.
Exploration of effector pathways
The contributions of prototypical inflammatory pathways to PM-induced responses have been studied widely in models of FDE-PM exposure. Both aromatic and polar fractions of FDE-PM have been shown to induce nuclear receptor factor 2 (Nrf2)-dependent heme oxygenase-1 (HO-1) expression, activation of mitogen-activated protein kinase (MAPK) pathways and induction of NFκB signalling in macrophages and epithelial cells [79, 80]. Similarly, repeat exposure to BDE-PM (produced through combustion of a FD-BD blend) promoted oxidative stress and inflammation in mice, as evidenced by increased concentrations of macrophages in the lungs, and heightened expression of TNF-α, NFκB, Nrf2 and HO-1 proteins [81]. Additionally, it was shown that PM produced through combustion of 20% blends of waste-grease biodiesel (WGPM) stimulated greater extracellular signal-regulated kinase (ERK)1/2 phosphorylation in BEAS-2B cells than FDE-PM [71]. These studies indicate that BDE-PM-induced responses share features with other PM-induced responses in redox sensitive signally pathways, as demonstrated through ERK1/2 activation, can be stimulated with differential potency compared with FDE-PM [71].
Focus has also moved towards characterisation of the mechanisms by which BD-induced pathways are stimulated in the lung. PM of varying composition have been shown to induce airway inflammation via activation of toll-like receptors (TLR); pattern recognition receptors that stimulate cytokine secretion, via NFκB, activating protein-1 and MAPK driven pathways [82]. FDE-PM have been shown to induce cytokine secretion and neutrophilia in a TLR4-dependent manner [83], but composition-dependent differences (including metal and endotoxin content) have been observed in the range of TLR species with which particles interact [84], indicating that this mechanism may differ for BDE-PM. Indeed, Traviss et al. demonstrated that WGPM exposure increased expression of TLR2, but not TLR4 in THP-1 macrophage lysates while FDE-PM displayed little association with TLR2 expression [71].
In addition, combustion products have been shown to trigger pulmonary responses by activating dormant cytosolic AhRs. Once bound by PAHs from the surface of combustion particles, AhRs undergo a conformational change that permits their translocation into the nucleus. In terms of PM toxicity, a widely explored outcome of this activity is induction of cytochrome P450 (CYP) enzymes, which play a critical role in the oxidative metabolism of lipophilic compounds, including PAHs themselves. While chemically inert in their environmental form, PAHs display considerable redox activity once oxidised by CYPs, promoting development of oxidative stress and mutagenicity [85].
Unfiltered combustion products of a B99 blend of BD have been shown to significantly upregulate CYP1A1 expression in human bronchial epithelial cells after 60 min of exposure to fresh, pure exhaust. Unfiltered FD combustion products also enhanced CYP1A1 expression, producing a statistically significant (but numerically comparable) increase at a faster rate than the BDE (20 min). For both fuel types, emissions collected in the presence of diesel particulate filters induced considerably greater CYP1A1 expression than their unfiltered counterparts, demonstrating that the gas and semi-volatile components of both BDE and FDE contribute greatly to their toxicity [86] Many in vitro studies of exhaust toxicity (and indeed, technical assessments of engine emissions) employ particle mass, number or surface area as a means of determining exposure dosages, with surface area suggested to be most relevant for assessments of pulmonary inflammation [87]. However, the evidence that volatile exhaust components can regulate critical aspects of the pulmonary response to PAH exposure indicates that particle-focused dose metrics may not represent the exposure-response relationship for total exhaust very well.
The effects that feedstock compositions have on BDE-induced CYP1A1 expression have also been explored in bronchial epithelial cells, demonstrating that PM produced from a blend of 13% HVO and 7% FAME in FD induces CYP1A1 expression to a greater extent than PM produced through combustion of non-supplemented FAME and FD blends (both 7 and 20%) after both 4 and 20 h of exposure [88]. This trend was observed consistently across a dose range of 10–100 μg/ml, but occurred in contrast to total PAH contents for the particles, indicating that specific PAH combinations or non-PAH components contributed to CYP1A1 expression in this model.
Focusing further on mechanisms of BD-induced CYP activation, H441 human pneumocytes were used to investigate the potential involvement of transient receptor potential ankyrin 1 (TRPA1) ion channels (mechanistic contributors to PM-induced disruption of calcium homeostasis and downstream cytokine secretion). The data showed that while CYP1A1/1B1 over-expression was induced BDE-PM in a dose-dependent manner (and with far greater potency than with FDE-PM), it was not attenuated by TRPA1 antagonists. While this indicated a lack of relationship between TRPA1 activity and CYP expression, IL-8 secretion did decrease significantly (~ 60%) in the presence of the antagonist, highlighting a role for TRPA1 in stimulation of BDE-PM-induced inflammatory cascades [89].
Progression towards a global approach
Cytokine arrays
By using molecular profiling methodologies, it is possible to obtain data with both qualitative and quantitative properties. Thus, through the assessment of large panels of variables, molecular interactions can be explored as part of a wider network, to determine which molecules are induced, or inhibited across the time-course of a response. Given that development and resolution of pulmonary inflammation are regulated by the complex interplay of pro and anti-inflammatory mediators, application of profiling approaches to the context of BDE exposure was led by the performance of cytokine arrays (summarised in Table 1). While continuing to compare the effects of exposure to BDE with those of FD emissions, these studies enabled novel hypotheses to be drawn regarding fuel-specific mechanisms of inflammatory cell recruitment to the lungs, thus offering policymakers an additional layer of evidence to support or oppose BD usage.
Performing an early, small-scale cytometric array on lung tissue, Shvedova et al. observed differential patterns of cytokine expression in the lungs of mice exposed to 0–500 μg/m3 concentrations of SME exhaust or FDE for a 4-week period [90]. The authors noted that BDE stimulated MCP-1 secretion by 38% more than FDE and that this response was consistent with Finch et al.’s observation that repeated exposure to BDE stimulated macrophage recruitment to rodent lungs [50]. As this response occurred in parallel to increased expression of IL-12p40, in the absence of an IFNγ, response with BDE exposure, the group hypothesised that BDE (but not necessarily FDE), induced macrophage activation via non-classical pathways. They also demonstrated that both exposure types induced increases in cytokine accumulation in the liver but that these effects were less pronounced than those observed in the lungs [90].
Several larger array platforms have been adopted to explore BDE and FDE toxicity, offering the distinct advantage of a more comprehensive qualitative range. Indeed, Yanamala et al. detected significant elevations of 17 cytokines and chemokines in murine pulmonary tissue following acute exposure to an 18 μg/mouse bolus of FAME exhaust PM (B100); a signature that translated closely to bronchoalveolar lavage fluid (BALF) [65] and was concordant with the induction of IL-12p70, IL-6, MCP-1 and TNFα secretion that was reported by Shvedova et al’s smaller scale SME study (Table 1). Yanamala et al. also observed evidence that BDE-PM induced secretion of ten cytokines and chemokines acutely, whilst FDE-PM did not [65]. This indicated that BDE-PM induces a more complex inflammatory cascade than FDE-PM; with the authors suggesting potential contributions of allergic inflammation and type 2 T helper cell responses [65]. Furthermore, the study demonstrated that IL-1a and IL-12p40 concentrations remained elevated 7 and 28 days after inoculation with BDE-PM, but not FDE-PM, indicating that BDE promotes a more prolonged pulmonary response [65].
Fukagawa et al. [29] used cytokine arrays to explore the effects of exposure to exhaust from commercially relevant B20 BD blends. Analysing the pulmonary tissue and BALF of mice, the authors demonstrated that repeated exposure to combustion particles from B20 SME (84 μg/ treatment over 3 days) induced pulmonary secretion of G-CSF, IP-10 and IL-6. While variations in study design and sample collection prevent a simple comparison of these results with those of Shvedova et al.’s murine study of exposure to PM from 100% SME [90], the authors did demonstrate that FDE-PM did not alter pulmonary cytokine profiles when compared with controls, despite inducing comparable BALF cell profiles to the B20 sample. This observation illustrates the value of board-based molecular investigations to comprehensively characterise the impacts that BDE-specific components have on pulmonary cells and is supported by the group’s observations in vitro. Using THP-1 and BEAS-2B cell lines, the group demonstrated that B20 BDE-PM and FDE-PM induce differential responses to one another in both macrophages and bronchial epithelial cells. A major concern for assessments of pollutant inflammogenicity is the ability of carbonaceous particles to bind cytokines, reducing the effective concentration during processing, thus risking introduction of type II errors during cytokine/chemokine quantifications [91]. Importantly, Fukagawa et al. used a cell-free system to demonstrate that neither particle species altered the concentration of cytokine standards, indicating that the observations were products of true interactions between the particles and the cells [29] rather than artefacts of differential particle composition.
Evidence that the physicochemical properties of particulates influence their toxicity was reported by Malorni et al. who demonstrated that particles with an aerodynamic diameter of less than 10 nm from either B20 FAME or pure FD emissions (1.2 or 4 ppm) inhibited cytokine secretion globally in A549 epithelial cells, [92]. Based on data collected for 27 targets (Table 1), the authors suggest that the small size of the particles was responsible for this, facilitating interference with mechanisms of protein synthesis or secretion, alternatively, that the assay had captured negative feedback loops following initial elevations in cytokine secretion [92]. It should however be noted that A549 express unusually high levels of HO-1 and Nrf2 [93] and that cells cultured under standard laboratory conditions experience a high basal layer of oxidative stress [94] so may respond differentially to oxidative stimuli compared with cells in vivo. Once again, the authors report biodiesel-specific alterations to the cellular cytokine profile.
Transcriptomics
To characterise a mild inflammatory response in the lungs of rats exposed to 20% blends of RME BDE for 7 or 28 days, the expression of 32 genes in pulmonary tissue was measured [95]. This semi-targeted approach drew upon the multivariate nature of transcriptomic analysis, but focused only on genes that are known to encode mediators of inflammatory pathways (including cytokines, antioxidants and xenobiotic metabolising enzymes) limiting the potential of the study to generate new hypotheses regarding exposure-induced response mechanisms. Indeed, of the four significant transcriptional changes observed, two (dysregulation of superoxide dismutase and glutathione peroxidase expression) have been associated with BDE exposure previously [61, 96]. In contrast, evidence that BDE induced expression of β-2 adrenergic receptors (β2AR) [95] was novel. Given that PM2.5 has been shown to induce IL-6 secretion in macrophages by stimulating β2ARs [97], this result may encourage innovative research into ion channel activity during BDE-induced inflammatory responses.
The first genome-wide transcriptomic analysis of BDE-induced toxicity helped resolve the difficulties in inter-study comparison that surrounded previous hypothesis-driven analyses. To detect mechanisms of toxicity that are common to different types of exhaust particulate, Libalova et al. used standardised methods of particle collection and cellular exposure to study global changes in bronchial epithelial cell gene expression following exposure to the organic fractions of exhaust particles produced through combustion of RME (100 and 30% blends), FD or pure NEXBTL (a combination of HVO and waste animal fat) PM. Due to the genome-wide scope of the array (performed using Illumina Human-HT12 v4 Expression BeadChips), the group were able to demonstrate that RME-derived organic matter altered expression of more genes than samples obtained from NEXBTL and that the extent of transcriptomic changes heightened with increasing RME:FD ratios in the parent fuels (from 0 to 100%) after 4 h exposure. Additionally, the experiment captured the transient changes in gene expression profiles during acute responses to particle extracts. After 4 h of exposure, many transcriptomic changes were induced in common by each of the tested PM fractions, influencing antioxidant defences, xenobiotic/ lipid metabolism, suppression of apoptotic stimuli and blood clotting. After 24 h however, far fewer genes and pathways were dysregulated by all particles and a greater number of fuel-specific dysregulated genes were observed (especially for the 30% RME blend). Most of these fuel-specific changes associated with NEXTBL exposure, indicating that PM produced by combustion of this BD altered expression of genes required to regulate cell cycle progression [98].
Similarly, RME emissions were shown to alter the expression of genes required for cell cycle progression in mice [61]. Pathway analysis (using Ingenuity Pathway Analysis software (IPA)) also associated exposure to RME and FD emissions with ‘metabolic disease, neurological disease, psychological disorders’. However, as discussed by the authors, IPA analyses depend upon the size of feature lists to estimate network scores and may not provide accurate representations of dysregulated pathways. Indeed, analyses performed with Pathway Studio software offered considerably different results, associating exposure to FD (but not RME) emissions with dysregulation of genes required for DNA repair pathways. This output contrasted with that produced by IPA but was supported by evidence that FDE induced development of acrolein adducts and induction of phosphorylated histone H2AX genotoxicity markers [61], highlighting the importance of selecting appropriate bioinformatic workflows.
It must be noted that molecular responses to particulate insult can occur downstream and independently of the transcriptome. For example, residual oily fly ash, urban dust and volcanic ash directly inhibit the activity of antioxidant enzymes with capacities that reflect their relative toxicity in vitro [64]. Mechanisms such as these would be excluded from characterisations of particle toxicity if transcriptomic screens were studied alone. Additionally, many ambient particulate species induce degradation of oxidised proteins [99] indicating that not all transcriptional events will yield protein-level activity under conditions of particle-induced oxidative stress.
Metabolomics and mass-spectrometry based profiling
As the product of upstream molecular events, the metabolome is considered an important source of mechanistically relevant biomarkers for cellular activities that can be explored alongside the transcriptome or proteome. Compared with these analyses however, production of comprehensive metabolomic profiles is challenging. Properties such as polarity or volatility require molecular subsets to be quantified through distinctly optimised platforms and annotation of metabolites in the resulting spectra is far from complete. A multi-platform approach has been used to measure the changes that BDE exposure induces upon the pulmonary metabolome; combining 1H nuclear magnetic resonance spectroscopy (1H NMR), liquid chromatography-mass spectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS) methods to identify markers of BDE toxicity in human bronchial wash (BW) and BALF samples [100]. By profiling distinct molecular subsets, this use of complimentary methods greatly enhanced the number of metabolites that were detected in the samples; a feature that related directly to the number of pathways that were associated with BDE exposure via pathway enrichment analysis. While requiring validation, connections between these pathways and BDE exposure were novel, implicating degradation of cell membrane lipids as a driver of the response alongside alterations in energy metabolism [100]. Importantly, the analysis demonstrated that BDE-induced alterations to pulmonary metabolism were highly compartment specific, with concentrations of individual metabolites differing significantly between the BW and BALF and a stronger response being detectable for peripheral regions of the lungs [100]. Extending this work, Gouveia-Fugueira et al. showed that human BW and BALF samples could be differentiated by their lipid mediator profiles with concentrations of prostaglandin- E2, 12,13-dihydroxy-9Z-octadecenoic acid, and 13-hydroxyoctadecadienoic acid being significantly elevated in BALF following exposure to BDE [101]. Given that these metabolites and inflammatory lipids are the products, substrates, intermediates and regulators of cellular reactions, this evidence provides a sturdy rationale for focusing on responses in the peripheral airways – an assumption that is often made based on estimated deposition patterns for similarly sized particles [102].
Future directions
Performance of omics-led experimentation has considerably advanced characterisations of the mechanisms that underlie BDE-induced toxicity (Fig. 2). Indeed, with the large range of platforms available for molecular profiling, it will be possible to identify novel pathways and regulators of toxicity and to make comprehensive comparisons between the molecular interactions that associate with BDE and FDE. As a starting point, expansion of metabonomic studies may be especially useful; as not only do metabolites incorporate the series of molecular events that occurred upstream of their synthesis (including epigenomic control of gene expression, post-translational modifications etc) but, as products of enzymatic reactions, they demonstrate those molecular events which have already occurred in samples. Subsequently, genomic, transcriptomic or proteomic data would be greatly useful further along the characterisation process where information regarding the origin of pathway dysregulation is required, though significant computational and statistical challenges exist in bring these large data sets together. Here, additional screens such as epigenetic or microRNA profiling will provide complementary information regarding the mechanisms by which gene transcription or protein synthesis are dysregulated by exposure.

General overview of known molecular and cellular features of pulmonary responses to BDE exposure, including details detected via targeted (a) and untargeted (b) experimental approaches in samples harvested from human cohorts or in vitro and in vivo models of human and rodent airways
Although availability of human lung tissue is limited by the invasive nature of biopsy, the multi-platform metabolomic screens performed have demonstrated that metabolite markers of pulmonary responses to BDE exposure are detectable outside of the tissue itself [100]. While collection of BALF or BW remains an invasive procedure, urine and plasma (both rich in metabolite species) can be harvested quickly and painlessly, promoting greater participant recruitment for studies. Metabolite biomarkers of inflammatory and oxidative stress responses to cigarette smoke and metallic welding fumes have been detected previously in human urine and blood [103, 104], indicating that these samples have potential as sources of mechanistically relevant biomarkers for pulmonary responses to other particulates. Should similar urinary or blood biomarkers be identified following BDE exposure, adverse outcomes of exposure to the pollutant could be detected and monitored in large human cohorts, enhancing the representability of characterised mechanisms to the population in general and permitting detection or exploration of the response in high susceptibility groups, including those with existing pulmonary conditions where non-critical, invasive procedures are ill advised [105].
Integration of omics approaches to encompass multiple molecular species could expand the number of pathways that are bioinformatically associated with exposure by generating a greater number of novel hypotheses. Indeed, the detection rate of mechanistically relevant pathways resulting from responses to chemotherapeutics was improved by 76% by integrating transcriptomic and metabolomic features during pathway analysis [106]. Considering that the incidence of type I errors increases with the number of variables that are input into a pathway analysis, complementary screens also provide the important function of cross-validation. The observation of a hierarchical proteomic response to FDE-PM exposure for example, has been validated at the transcriptional level by evidence that mid-range concentrations of FDE-PM induce expression of genes that encode pro-inflammatory mediators but not antioxidant enzymes in rat macrophages [107].
Finally, it would be prudent to apply profiling techniques to the exploration of non-classical models of pulmonary exposure, widening our understanding beyond responses of the airway epithelium and innate immune system. For example, possible neuronal responses to BDE exposure were highlighted by transcriptomic analysis of mouse lung (associating RME emission exposure with ‘neuronal disease’ pathways) and the discovery that the SOF of BDE-PM agonises TRPA1 activity [89]. Comprehensive studies of molecular alterations within pulmonary neurons could greatly enhance the breadth of our characterisations of BDE-induced pulmonary toxicity, potentially providing novel explanations of how exposure may trigger respiratory reflexes.
Conclusions
Introduction of omics strategies to the characterisation of toxicity pathways has greatly progressed our understanding of how human lungs respond to BDE. Moving away from hypotheses inspired by our knowledge of how the lungs respond to FDE, profiling methods have identified mechanistically informative, fuel-specific molecular signatures. Importantly, they have also highlighted mechanisms beyond those that we traditionally associate with particulate exposure, which could encourage exploration into the roles of additional pathways and physiological systems. Such outcomes are difficult to achieve using traditional toxicological testing but would be highly beneficial throughout the field of particulate toxicology. The most recent air quality proposals (such as the UK Clean Air Strategy 2018) pledge to address a range of pollutant sources much wider than the transport combustion engine [108]. Characterisations of the toxicity posed by many of these particles are in their infancy but, with the breadth of information that omics screens offer, policy makers could be provided with a more detailed understanding of how the different particles impact the lungs, and whether some cause more diverse toxicities than others. This information could assist targeting of regulatory efforts to those pollutants that pose the greatest risk to public health and help to prevent unexpected consequences by focusing on new fuels, or engine technologies that may offer solutions to current problems but introduce a new set of problems. The history of air pollution regulation is littered with such examples [109], with the European policies of the mid-1990s promoting the uptake of diesel vehicles being the clearest contemporary example. Embracing the advances in multi-omics technologies, allied to traditional hypothesis-led approaches to address the safety of new fuels presents the opportunity to develop evidence-based pollutant mitigations strategies, rather than simplistic fixes that often fail to consider the health of the population.
Abbreviations
- 1H NMR:
-
Proton nuclear magnetic resonance spectroscopy
- AhR:
-
Aryl-hydrocarbon receptor
- BALF:
-
Bronchoalveolar lavage fluid
- BDE:
-
Biodiesel exhaust
- BDE-PM:
-
Biodiesel exhaust particulate matter
- bFGF:
-
Basic fibroblast growth factor
- BW:
-
Bronchial wash
- CCL:
-
Chemokine (C-C-motif) ligand
- CO:
-
Carbon monoxide
- CXCL:
-
Chemokine (C-X-motif) ligand
- CYP:
-
Cytochrome P450
- EGFR:
-
Epidermal growth factor receptor
- EPA:
-
Environmental Protection Agency
- ERK:
-
Extracellular signal-regulated kinase
- FAME:
-
Fatty acid methyl ester
- FD:
-
Fossil diesel
- FDE:
-
Fossil diesel exhaust
- FDE-PM:
-
Fossil diesel exhaust particulate matter
- GC-MS:
-
Gas chromatography-mass spectrometry
- G-CSF:
-
Granulocyte-colony stimulating factor
- HO-1:
-
Heme oxygenase-1
- HVO:
-
Hydrogenated vegetable oil
- IL:
-
Interleukin
- IP-10:
-
Interferon-γ induced protein
- IPA:
-
Ingenuity Pathway Analysis
- LC-MS:
-
Liquid chromatography-mass spectrometry
- LIF:
-
Leukaemia inhibitory factor
- MAOK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemoattractant protein-1
- M-CSF:
-
Macrophage colony stimulating factor
- MIP:
-
Macrophage inflammatory protein
- NFκB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NIFNγ:
-
Interferon-γ neutralising
- NOx :
-
Nitrogen oxides
- Nrf2:
-
Nuclear receptor factor 2
- PAH:
-
Polycyclic aromatic hydrocarbon
- PDGF:
-
Platelet-derived growth factor
- PM:
-
Particulate matter
- PM0.1 :
-
Ultrafine particulates
- RME:
-
Rapeseed methyl ester
- SME:
-
Soybean methyl ester
- SOF:
-
Soluble organic fraction
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumour necrosis factor-α
- TRPA1:
-
Transient receptor potential ankyrin 1
- VEGF:
-
Vascular endothelial growth factor
- WGPM:
-
Waste grease biodiesel exhaust particulate matter
- β2AR:
-
β-2 adrenergic receptors
References
Department for Transport UK. Provisional estimates show rolling annual motor vehicle traffic has increased to a record high for the ninth consecutive quarter [Internet]. London; 2017 [cited 2018 Feb 15]. Available from: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/663531/prov-road-traffic-estimates-oct-2016-to-sep-2017.pdf.
Department for Transport UK. Road Traffic Forecasts: 2015 [Internet]. London; 2015. Available from: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/411471/road-traffic-forecasts-2015.pdf.
Van Essen H, Delft CE. Global Forum on Transport and Environment in a Globalising World The Environmental Impacts of Increased International Road and Rail Freight Transport Past trends and future perspectives [Internet]. [cited 2018 Nov 8]. Available from: http://www.oecd.org/greengrowth/greening-transport/41380980.pdf
Pant P, Harrison RM. Estimation of the contribution of road traffic emissions to particulate matter concentrations from field measurements: A review. Atmos Environ. 2013;77:78–97. [cited 2018 Feb 15]. Available from: https://www.sciencedirect.com/science/article/pii/S1352231013002732
Garg BD. Brake wear particulate matter emissions. 2000.
Kupiainen KJ, Tervahattu H, Räisänen M, Mäkelä T, Aurela M, Hillamo R. Size and composition of airborne particles from pavement Wear, tires, and traction sanding. Environ Sci Technol. 2005;39(3):699–706 Available from: https://doi.org/10.1021/es035419e.
Department of Health and Human Services. Diesel Exhaust Particles [Internet]. Report on Carcinogens, 14th Edition. 2000 [cited 2017 Apr 18]. Available from: https://ntp.niehs.nih.gov/ntp/roc/content/profiles/dieselexhaustparticulates.pdf
Iskandar A, Andersen ZJ, Bønnelykke K, Ellermann T, Andersen KK, Bisgaard H. Coarse and fine particles but not ultrafine particles in urban air trigger hospital admission for asthma in children. Thorax. 2012;67(3):252 Available from: http://thorax.bmj.com/content/67/3/252.abstract.
Ostro B, Roth L, Malig B, Marty M. The effects of fine particle components on respiratory hospital admissions in children. Environ Health Perspect. 2009;117:475–80.
Vichit-Vadakan N, Ostro BD, Chestnut LG, Mills DM, Aekplakorn W, Wangwongwatana S, et al. Air pollution and respiratory symptoms: results from three panel studies in Bangkok, Thailand. Environ Health Perspect [Internet]. 2001;109(Suppl 3):381–7 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1240555/.
Zemp E, Elsasser S, Schindler C, Künzli N, Perruchoud AP, Domenighetti G, et al. Long-Term Ambient Air Pollution and Respiratory Symptoms in Adults (SAPALDIA Study). Am J Respir Crit Care Med. 1999 159(4):1257–1266. [cited 2017 Jul 22]. Available from: http://www.atsjournals.org/doi/abs/10.1164/ajrccm.159.4.9807052
Schwab AW, Bagby MO, Freedman B. Preparation and properties of diesel fuels from vegetable oils. [cited 2018 Feb 15]; Available from: https://ac.els-cdn.com/0016236187901840/1-s2.0-0016236187901840-main.pdf?_tid=03cad016-1236-11e8-ac2e-00000aacb362&acdnat=1518688563_867e940db05f834ddad53b2fa5b99e6f
Balat M, Balat H. Progress in biodiesel processing. Appl Energy. 2010;87(6):1815–1835. [cited 2018 Feb 15]. Available from: https://www.sciencedirect.com/science/article/pii/S0306261910000255
Senatore A, Cardone M, Rocco V, Prati MV. A Comparative Analysis of Combustion Process in D.I. Diesel Engine Fueled with Biodiesel and Diesel Fuel. In 2000 [cited 2018 Feb 15]. Available from: http://papers.sae.org/2000-01-0691/
Morris RE, Pollack AK, Mansell GE, Lindhjem C, Jia Y, Wilson G. Impact of Biodiesel Fuels on Air Quality and Human Health: Summary Report; September 16, 1999--January 31, 2003 [Internet]. Golden, CO; 2003 May [cited 2018 Feb 15]. Available from: http://www.osti.gov/servlets/purl/15003890-Y3wSun/native/
Demirbas A. Importance of biodiesel as transportation fuel. Energy Policy. 2007;35(9):4661–4670. [cited 2018 Feb 15]. Available from: https://www.sciencedirect.com/science/article/pii/S0301421507001516
Jedrychowski W, Galas A, Pac A, Flak E, Camman D, Rauh V, et al. Prenatal Ambient Air Exposure to Polycyclic Aromatic Hydrocarbons and the Occurrence of Respiratory Symptoms over the First Year of Life. Eur J Epidemiol. 2005;20(9):775–782. [cited 2018 Nov 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16170661.
Gale SL, Noth EM, Mann J, Balmes J, Hammond SK, Tager IB. Polycyclic aromatic hydrocarbon exposure and wheeze in a cohort of children with asthma in Fresno, CA. J Expo Sci Environ Epidemiol. 2012;22(4):386–392. [cited 2018 Nov 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22549720.
Schwartz J, Slater D, Larson T V, Pierson WE, Koenig JQ. Particulate Air Pollution and Hospital Emergency Room Visits for Asthma in Seattle. Am Rev Respir Dis [Internet]. 1993;147(4):826–831. Available from: https://doi.org/10.1164/ajrccm/147.4.826
Anderson HR, Bremner SA, Atkinson RW, Harrison RM, Walters S. Particulate matter and daily mortality and hospital admissions in the west midlands conurbation of the United Kingdom: associations with fine and coarse particles, black smoke and sulphate. [cited 2018 Nov 8]; Available from: http://oem.bmj.com/
The european parliament and the council of the european union. directive 2009/28/ec of the european parliament and of the council on the promotion of the use of energy from renewable sources and amending and subsequently repealing directives 2001/77/EC and 2003/30/EC [Internet]. 2009. Available from: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:32009L0028
Kim D-S, Hanifzadeh M, Kumar A. Trend of Biodiesel feedstock and its impact onBiodiesel emission characteristics. Environ Prog Sustaiable Energy [Internet]. 2018;37(1):7–18 Available from: https://onlinelibrary.wiley.com/doi/epdf/10.1002/ep.12800.
Kim BG, Lee PH, Lee SH, Kim YE, Shin MY, Kang Y, et al. Long-Term Effects of Diesel Exhaust Particles on Airway Inflammation and Remodeling in a Mouse Model. Allergy Asthma Immunol Res. 2016;8(3):246–256. [cited 2017 Jul 1]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26922935.
Oberdörster G, Celein RM, Ferin J, Weiss B. Association of Particulate Air Pollution and Acute Mortality: Involvement of Ultrafine Particles? Inhal Toxicol. 1995;7(1):111–124. [cited 2018 Apr 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11541043.
Brown DM, Wilson MR, MacNee W, Stone V, Donaldson K. Size-dependent proinflammatory effects of ultrafine polystyrene particles: a role for surface area and oxidative stress in the enhanced activity of ultrafines. Toxicol Appl Pharmacol. 2001;175 Available from: https://doi.org/10.1006/taap.2001.9240.
César ACG, Carvalho JA, Nascimento LFC, Nascimento LFC. Association between NOx exposure and deaths caused by respiratory diseases in a medium-sized Brazilian city. Brazilian J Med Biol Res. 2015;48(12):1130–1135. [cited 2018 Apr 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26421866.
Wichmann HE, Spix C, Tuch T, Wölke G, Peters A, Heinrich J, et al. Daily mortality and fine and ultrafine particles in Erfurt, Germany part I: role of particle number and particle mass. Res Rep Health Eff Inst. 2000;(98):5–86-94. [cited 2018 Apr 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11918089.
Machado Corrêa S, Arbilla G. Carbonyl emissions in diesel and biodiesel exhaust. Atmos Environ. 2008;42(4):769–775. [cited 2018 Nov 6]. Available from: https://www.sciencedirect.com/science/article/pii/S1352231007008746
Fukagawa NK, Li M, Poynter ME, Palmer BC, Parker E, Kasumba J, et al. Soy Biodiesel and Petrodiesel Emissions Differ in Size, Chemical Composition and Stimulation of Inflammatory Responses in Cells and Animals. Environ Sci Technol. 2013;47(21):12496–12504. [cited 2018 Feb 21]. Available from: http://pubs.acs.org/doi/10.1021/es403146c
Xiao GG, Wang M, Li N, Loo JA, Nel AE. Use of Proteomics to Demonstrate a Hierarchical Oxidative Stress Response to Diesel Exhaust Particle Chemicals in a Macrophage Cell Line. J Biol Chem. 2003;278(50):50781–50790. [cited 2017 mar 6]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14522998.
Verheyen GR, Nuijten J-M, Van Hummelen P, Schoeters GR. Microarray analysis of the effect of diesel exhaust particles on in vitro cultured macrophages. Toxicol Vitr. 2004;18(3):377–391. [cited 2017 Mar 6]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0887233303002170
Bünger J, Krahl J, Baum K, Schröder O, Müller M, Westphal G, et al. Cytotoxic and mutagenic effects, particle size and concentration analysis of diesel engine emissions using biodiesel and petrol diesel as fuel. Arch Toxicol. 2000;74(8):490–498. [cited 2018 Feb 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11097388.
Madden MC. A paler shade of green? The toxicology of biodiesel emissions: Recent findings from studies with this alternative fuel. Biochim Biophys Acta. 2016;1860(12):2856–2862. [cited 2018 Oct 30]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27261091.
Brimblecombe P. History of Urban Air Pollution. In Springer, Dordrecht; 1998 . 7–20. [cited 2018 Feb 16]. Available from: http://link.springer.com/10.1007/978-94-015-9080-8_2
Salvi S, Blomberg A, Rudell B, Kelly F, Sandström T, Holgate ST, et al. Acute Inflammatory Responses in the Airways and Peripheral Blood After Short-Term Exposure to Diesel Exhaust in Healthy Human Volunteers. Am J Respir Crit Care Med. 1999;159(3):702–709. [cited 2017 Mar 7]. Available from: http://www.atsjournals.org/doi/abs/10.1164/ajrccm.159.3.9709083
Behndig AF, Larsson N, Brown JL, Stenfors N, Helleday R, Duggan ST, et al. Proinflammatory doses of diesel exhaust in healthy subjects fail to elicit equivalent or augmented airway inflammation in subjects with asthma. Thorax. 2011;66(1):12–9. [cited 2018 Nov 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20837873.
Steenhof M, Mudway IS, Gosens I, Hoek G, Godri KJ, Kelly FJ, et al. Acute nasal pro-inflammatory response to air pollution depends on characteristics other than particle mass concentration or oxidative potential: the RAPTES project. Occup Environ Med. 2013;70(5):341–348. [cited 2018 Nov 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23428835.
Strak M, Janssen NAH, Godri KJ, Gosens I, Mudway IS, Cassee FR, et al. Respiratory health effects of airborne particulate matter: the role of particle size, composition, and oxidative potential-the RAPTES project. Environ Health Perspect. 2012;120(8):1183–1189. [cited 2018 Nov 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22552951.
McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, et al. Respiratory Effects of Exposure to Diesel Traffic in Persons with Asthma. N Engl J Med. 2007;357(23):2348–2358. [cited 2018 Nov 8]. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMoa071535
Janssen NAH, Hoek G, Simic-Lawson M, Fischer P, van Bree L, ten Brink H, et al. Black Carbon as an Additional Indicator of the Adverse Health Effects of Airborne Particles Compared with PM10 and PM2.5. Environ Health Perspect. 2011;119(12):1691–1699. [cited 2018 Aug 15]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21810552.
Atkinson RW, Analitis A, Samoli E, Fuller GW, Green DC, Mudway IS, et al. Short-term exposure to traffic-related air pollution and daily mortality in London, UK. J Expo Sci Environ Epidemiol. 2016;26(2):125–132. [cited 2018 Aug 15]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26464095.
Peng RD, Bell ML, Geyh AS, McDermott A, Zeger SL, Samet JM, et al. Emergency Admissions for Cardiovascular and Respiratory Diseases and the Chemical Composition of Fine Particle Air Pollution. Environ Health Perspect. 2009;117(6):957–963. [cited 2018 Aug 15]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19590690.
Stenfors N, Nordenhäll C, Salvi SS, Mudway I, Söderberg M, Blomberg A, et al. Different airway inflammatory responses in asthmatic and healthy humans exposed to diesel. Eur Respir J. 2004 Jan;23(1):82–6.
Rudell B, Blomberg A, Helleday R, Ledin MC, Lundbäck B, Stjernberg N, et al. Bronchoalveolar inflammation after exposure to diesel exhaust: comparison between unfiltered and particle trap filtered exhaust. Occup Environ Med. 1999;56(8):527–534. [cited 2018 Nov 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10492649.
Pourazar J, Mudway IS, Samet JM, Helleday R, Blomberg A, Wilson SJ, et al. Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. Am J Physiol - Lung Cell Mol Physiol [Internet]. 2005 [cited 2017 Sep 14];289(5). Available from: http://ajplung.physiology.org/content/289/5/L724.long
Robinson RK, Birrell MA, Adcock JJ, Wortley MA, Dubuis ED, Chen S, et al. Mechanistic link between diesel exhaust particles and respiratory reflexes. J Allergy Clin Immunol [Internet]. 2018 Mar 1 [cited 2018 May 1];141(3):1074–1084.e9. Available from: https://www.sciencedirect.com/science/article/pii/S0091674917307960
Carlsten C, Blomberg A, Pui M, Sandstrom T, Wong SW, Alexis N, et al. Diesel exhaust augments allergen-induced lower airway inflammation in allergic individuals: a controlled human exposure study. Thorax [Internet]. 2016 Jan [cited 2018 Nov 8];71(1):35–44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26574583.
Hosseini A, Hirota JA, Hackett TL, McNagny KM, Wilson SJ, Carlsten C. Morphometric analysis of inflammation in bronchial biopsies following exposure to inhaled diesel exhaust and allergen challenge in atopic subjects. Part Fibre Toxicol [Internet]. 2015 Dec 13 [cited 2018 Nov 8];13(1):2. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26758251.
Holgate ST. “Every breath we take: the lifelong impact of air pollution” – a call for action. Clin Med (Northfield Il) [Internet]. 2017 Feb 1 [cited 2018 Nov 8];17(1):8–12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28148571.
Finch GL, Hobbs CH, Blair LF, Barr EB, Hahn FF, Jaramillo RJ, et al. Effects of subchronic inhalation exposure of rats to emissions from a diesel engine burning soybean oil-derived biodiesel fuel. Inhal Toxicol [Internet]. 2002 Jan [cited 2018 Feb 19];14(10):1017–1048. Available from: http://www.tandfonline.com/doi/full/10.1080/08958370290084764
Eckl PM, Leikermoser P, Wörgetter M, Prankl H, Wurst F. The Mutagenic Potential of Diesel and Biodiesel Exhausts. In: Plant Oils as Fuels [Internet]. Berlin, Heidelberg: Springer Berlin Heidelberg; 1998 [cited 2018 Feb 19]. p. 123–140. Available from: http://www.springerlink.com/index/10.1007/978-3-642-72269-1_10
Swanson KJ, Madden MC, Ghio AJ. Biodiesel exhaust: the need for health effects research. Environ Health Perspect [Internet]. 2007 Apr [cited 2018 Feb 19];115(4):496–499. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17450214.
Whittaker MH. Risk Assessment and Alternatives Assessment: Comparing Two Methodologies. Risk Anal [Internet]. 2015 Dec 1 [cited 2018 Feb 20];35(12):2129–2136. Available from: http://doi.wiley.com/10.1111/risa.12549
International Agency for Research on Cancer. Diesel and Gasoline Engine Exhausts and Some Nitroarenes [Internet]. 2014 [cited 2018 Nov 5]. Available from: https://monographs.iarc.fr/wp-content/uploads/2018/06/mono105.pdf
Carraro E, Locatelli AL, Ferrero C, Fea E, Gilli G. Biological activity of particle exhaust emissions from light-duty diesel engines. J Environ Pathol Toxicol Oncol [Internet]. 1997 [cited 2018 Nov 5];16(2–3):101–109. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9275990.
Ross JA, Nelson GB, Mutlu E, Warren SH, Gilmour MI, DeMarini DM. DNA adducts induced by in vitro activation of extracts of diesel and biodiesel exhaust particles. Inhal Toxicol [Internet]. 2015 Sep 19 [cited 2018 Oct 1];27(11):576–584. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26514785.
Topinka J, Milcova A, Schmuczerova J, Mazac M, Pechout M, Vojtisek-Lom M. Genotoxic potential of organic extracts from particle emissions of diesel and rapeseed oil powered engines. Toxicol Lett [Internet]. 2012 Jul 7 [cited 2018 Nov 5];212(1):11–17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22562013.
Jalava PI, Tapanainen M, Kuuspalo K, Markkanen A, Hakulinen P, Happo MS, et al. Toxicological effects of emission particles from fossil- and biodiesel-fueled diesel engine with and without DOC/POC catalytic converter. Inhal Toxicol [Internet]. 2010 Dec 29 [cited 2018 Feb 19];22(sup2):48–58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21029031.
Hemmingsen JG, Møller P, Nøjgaard JK, Roursgaard M, Loft S. Oxidative Stress, Genotoxicity, And Vascular Cell Adhesion Molecule Expression in Cells Exposed to Particulate Matter from Combustion of Conventional Diesel and Methyl Ester Biodiesel Blends. Environ Sci Technol [Internet]. 2011 Oct 1 [cited 2018 Nov 5];45(19):8545–8551. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21842833.
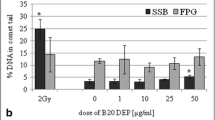
Cervena T, Rossnerova A, Sikorova J, Beranek V, Vojtisek-Lom M, Ciganek M, et al. DNA Damage Potential of Engine Emissions Measured In Vitro by Micronucleus Test in Human Bronchial Epithelial Cells. Basic Clin Pharmacol Toxicol [Internet]. 2017 Sep [cited 2018 Nov 5];121:102–108. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27782363.
Douki T, Corbière C, Preterre D, Martin PJ, Lecureur V, André V, et al. Comparative study of diesel and biodiesel exhausts on lung oxidative stress and genotoxicity in rats. Environ Pollut [Internet]. 2018 Apr 8 [cited 2018 Feb 5];235:514–524. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29324381.
Schwarze PE, Totlandsdal AI, Låg M, Refsnes M, Holme JA, Øvrevik J. Inflammation-related effects of diesel engine exhaust particles: studies on lung cells in vitro. Biomed Res Int [Internet]. 2013 Feb 14 [cited 2018 Feb 19];2013:685142. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23509760.
Swanson KJ, Kado NY, Funk WE, Pleil JD, Madden MC, Ghio AJ. Release of the Pro-Inflammatory Markers by BEAS-2B Cells Following In Vitro Exposure to Biodiesel Extracts. Open Toxicol J [Internet]. 2009 Mar 19 [cited 2018 Feb 19];3(1):8–15. Available from: http://benthamopen.com/ABSTRACT/TOTOXIJ-3-8
Gerlofs-Nijland ME, Totlandsdal AI, Tzamkiozis T, Leseman DLAC, Samaras Z, Låg M, et al. Cell Toxicity and Oxidative Potential of Engine Exhaust Particles: Impact of Using Particulate Filter or Biodiesel Fuel Blend. Environ Sci Technol [Internet]. 2013 Jun 4 [cited 2018 Feb 5];47(11):5931–5938. Available from: http://pubs.acs.org/doi/10.1021/es305330y
Yanamala N, Hatfield MK, Farcas MT, Schwegler-Berry D, Hummer JA, Shurin MR, et al. Biodiesel versus diesel exposure: Enhanced pulmonary inflammation, oxidative stress, and differential morphological changes in the mouse lung. Toxicol Appl Pharmacol [Internet]. 2013 Oct 15 [cited 2018 Jan 11];272(2):373–383. Available from: http://www.sciencedirect.com/science/article/pii/S0041008X13003098
Gavett SH, Wood CE, Williams MA, Cyphert JM, Boykin EH, Daniels MJ, et al. Soy biodiesel emissions have reduced inflammatory effects compared to diesel emissions in healthy and allergic mice. Inhal Toxicol [Internet]. 2015 Sep 19 [cited 2018 Oct 25];27(11):533–544. Available from: http://www.tandfonline.com/doi/full/10.3109/08958378.2015.1054966
Bass VL, Schladweiler MC, Nyska A, Thomas RF, Miller DB, Krantz T, et al. Comparative cardiopulmonary toxicity of exhausts from soy-based biofuels and diesel in healthy and hypertensive rats. Inhal Toxicol [Internet]. 2015 Sep 19 [cited 2018 Feb 20];27(11):545–556. Available from: http://www.tandfonline.com/doi/full/10.3109/08958378.2015.1060279
Wood HE, Marlin N, Mudway IS, Bremner SA, Cross L, Dundas I, et al. Effects of Air Pollution and the Introduction of the London Low Emission Zone on the Prevalence of Respiratory and Allergic Symptoms in Schoolchildren in East London: A Sequential Cross-Sectional Study. PLoS One [Internet]. 2015 Aug 21;10(8):e0109121-. Available from: http://dx.doi.org/10.1371/journal.pone.0109121.
Mullins BJ, Kicic A, Ling K-M, Mead-Hunter R, Larcombe AN. Biodiesel exhaust-induced cytotoxicity and proinflammatory mediator production in human airway epithelial cells. Environ Toxicol [Internet]. 2016 Jan [cited 2018 Feb 20];31(1):44–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25045158.
Liu Y-Y, Lin T-C, Wang Y-J, Ho W-L. Biological toxicities of emissions from an unmodified engine fueled with diesel and biodiesel blend. J Environ Sci Heal Part A [Internet]. 2008 Nov 5 [cited 2018 Feb 20];43(14):1735–1743. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18988112.
Traviss N, Li M, Lombard M, Thelen BA, Palmer BC, Poynter ME, et al. Petrodiesel and waste grease biodiesel (B20) emission particles at a rural recycling center: characterization and effects on lung epithelial cells and macrophages. Air Qual Atmos Heal [Internet]. 2014 Mar 9 [cited 2018 Feb 20];7(1):59–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29430261.
Betha R, Pavagadhi S, Sethu S, Hande MP, Balasubramanian R. Comparative in vitro cytotoxicity assessment of airborne particulate matter emitted from stationary engine fuelled with diesel and waste cooking oil-derived biodiesel. Atmos Environ [Internet]. 2012 Dec 1 [cited 2018 Feb 20];61:23–29. Available from: https://www.sciencedirect.com/science/article/pii/S135223101200670X
Kulkarni MG, Dalai AK. Waste Cooking OilsAn Economical Source for Biodiesel: A Review. [cited 2018 Feb 20]; Available from: https://pubs.acs.org/doi/pdf/10.1021/ie0510526
Oberdörster G, Ferin J, Soderholm S, Gelein R, Cox C, Baggs R, et al. Increased Pulmonary Toxicity of Inhaled Ultrafine Particles: Due to Lung Overload Alone? Ann Occup Hyg [Internet]. 1994 Jan 1 [cited 2018 Nov 2];38(inhaled_particles_VII):295–302. Available from: https://academic.oup.com/annweh/article/38/inhaled_particles_VII/295/208692/Increased-Pulmonary-Toxicity-of-Inhaled-Ultrafine
Stoeger T, Reinhard C, Takenaka S, Schroeppel A, Karg E, Ritter B, et al. Instillation of Six Different Ultrafine Carbon Particles Indicates a Surface Area Threshold Dose for Acute Lung Inflammation in Mice. Environ Health Perspect [Internet]. 2006 Mar [cited 2018 Nov 1];114(3):328–333. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16507453.
Osier M, Baggs RB, Oberdörster G. Intratracheal instillation versus intratracheal inhalation: influence of cytokines on inflammatory response. Environ Health Perspect [Internet]. 1997 Sep [cited 2017 Aug 9];105 Suppl 5:1265–1271. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9400736.
Warheit DB, Kreiling R, Levy LS. Relevance of the rat lung tumor response to particle overload for human risk assessment—Update and interpretation of new data since ILSI 2000. Toxicology [Internet]. 2016 Dec 30 [cited 2018 Nov 5];374:42–59. Available from: https://www.sciencedirect.com/science/article/pii/S0300483X1630292X
Sager TM, Castranova V. Surface area of particle administered versus mass in determining the pulmonary toxicity of ultrafine and fine carbon black: comparison to ultrafine titanium dioxide. Part Fibre Toxicol [Internet]. 2009 May 4 [cited 2018 Nov 2];6:15. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19413904.
Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol [Internet]. 2004 Sep 1 [cited 2018 Feb 22];173(5):3467–3481. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15322212.
Marano F, Boland S, Bonvallot V, Baulig A, Baeza-Squiban A. Human airway epithelial cells in culture for studying the molecular mechanisms of the in£ammatory response triggered by diesel exhaust particles. [cited 2018 Feb 22]; Available from: https://link.springer.com/content/pdf/10.1023%2FA%3A1019548517877.pdf
Valenca S dos S, Cattani-Cavalieri I, Carvalho GMC, Zin WA, Costa AMA, Porto LC, et al. Acute exposure to diesel-biodiesel particulate matter promotes murine lung oxidative stress by Nrf2/HO-1 and inflammation through the NF-κB/TNF-α pathways. Free Radic Biol Med [Internet]. 2017 Nov 1 [cited 2018 May 25];112:201. Available from: https://www.sciencedirect.com/science/article/pii/S0891584917311012
Bauer RN, Diaz-Sanchez D, Jaspers I. Effects of air pollutants on innate immunity: the role of Toll-like receptors and nucleotide-binding oligomerization domain-like receptors. J Allergy Clin Immunol [Internet]. 2012 Jan [cited 2018 Feb 22];129(1):14–24-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22196521.
Inoue K, Takano H, Yanagisawa R, Hirano S, Ichinose T, Shimada A, et al. The role of toll-like receptor 4 in airway inflammation induced by diesel exhaust particles. Arch Toxicol [Internet]. 2006;80(5):275–9 Available from: https://doi.org/10.1007/s00204-005-0040-6.
Shoenfelt J, Mitkus RJ, Zeisler R, Spatz RO, Powell J, Fenton MJ, et al. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J Leukoc Biol [Internet]. 2009 Aug 1 [cited 2017 mar 6];86(2):303–312. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19406832.
Moorthy B, Chu C, Carlin DJ. Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicol Sci [Internet]. 2015 May [cited 2018 Feb 16];145(1):5–15. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25911656.
Hawley B, L’Orange C, Olsen DB, Marchese AJ, Volckens J. Oxidative Stress and Aromatic Hydrocarbon Response of Human Bronchial Epithelial Cells Exposed to Petro- or Biodiesel Exhaust Treated with a Diesel Particulate Filter. Toxicol Sci [Internet]. 2014 Oct 1 [cited 2018 Feb 21];141(2):505–514. Available from: http://academic.oup.com/toxsci/article/141/2/505/2511607/Oxidative-Stress-and-Aromatic-Hydrocarbon-Response
Schmid O, Stoeger T. Surface area is the biologically most effective dose metric for acute nanoparticle toxicity in the lung. J Aerosol Sci [Internet]. 2016 Sep 1 [cited 2018 Nov 1];99:133–143. Available from: https://www.sciencedirect.com/science/article/pii/S0021850215301166
Skuland TS, Refsnes M, Magnusson P, Oczkowski M, Gromadzka-Ostrowska J, Kruszewski M, et al. Proinflammatory effects of diesel exhaust particles from moderate blend concentrations of 1st and 2nd generation biodiesel in BEAS-2B bronchial epithelial cells—The FuelHealth project. Environ Toxicol Pharmacol [Internet]. 2017 Jun [cited 2018 Feb 22];52:138–142. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1382668917301035
Jaramillo IC, Sturrock A, Ghiassi H, Woller DJ, Deering-Rice CE, Lighty JS, et al. Effects of fuel components and combustion particle physicochemical properties on toxicological responses of lung cells. J Environ Sci Heal Part A [Internet]. 2018 Mar 21 [cited 2018 Feb 21];53(4):295–309. Available from: https://www.tandfonline.com/doi/full/10.1080/10934529.2017.1400793
Shvedova AA, Yanamala N, Murray AR, Kisin ER, Khaliullin T, Hatfield MK, et al. Oxidative Stress, Inflammatory Biomarkers, and Toxicity in Mouse Lung and Liver after Inhalation Exposure to 100% Biodiesel or Petroleum Diesel Emissions. J Toxicol Environ Heal Part A [Internet]. 2013 Aug 3 [cited 2018 Jan 11];76(15):907–921. Available from: http://www.tandfonline.com/doi/abs/10.1080/15287394.2013.825217
Kocbach A, Totlandsdal AI, Låg M, Refsnes M, Schwarze PE. Differential binding of cytokines to environmentally relevant particles: A possible source for misinterpretation of in vitro results? Toxicol Lett [Internet]. 2008 Jan 30 [cited 2018 Oct 29];176(2):131–137. Available from: https://www.sciencedirect.com/science/article/pii/S037842740700999X
Malorni L, Guida V, Sirignano M, Genovese G, Petrarca C, Pedata P. Exposure to sub-10 nm particles emitted from a biodiesel-fueled diesel engine: In vitro toxicity and inflammatory potential. Toxicol Lett [Internet]. 2017 Mar 15 [cited 2018 Feb 21];270:51–61. Available from: https://www.sciencedirect.com/science/article/pii/S0378427417300577
Kweon M-H, Adhami VM, Lee J-S, Mukhtar H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J Biol Chem [Internet]. 2006 Nov 3 [cited 2017 Aug 10];281(44):33761–33772. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16950787.
Kumar A, Dailey LA, Swedrowska M, Siow R, Mann GE, Vizcay-Barrena G, et al. Quantifying the magnitude of the oxygen artefact inherent in culturing airway cells under atmospheric oxygen versus physiological levels. Halliwell B, editor. FEBS Lett [Internet]. 2016 Jan [cited 2018 Nov 9];590(2):258–269. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26823171.
Magnusson P, Oczkowski M, Øvrevik J, Gajewska M, Wilczak J, Biedrzycki J, et al. No adverse lung effects of 7- and 28-day inhalation exposure of rats to emissions from petrodiesel fuel containing 20% rapeseed methyl esters (B20) with and without particulate filter – the FuelHealth project. Inhal Toxicol [Internet]. 2017 Apr 16 [cited 2018 Mar 2];29(5):206–218. Available from: https://www.tandfonline.com/doi/full/10.1080/08958378.2017.1339149
Adenuga AA, Wright ME, Atkinson DB. Evaluation of the reactivity of exhaust from various biodiesel blends as a measure of possible oxidative effects: A concern for human exposure. Chemosphere [Internet]. 2016 Mar [cited 2018 mar 2];147:396–403. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26774305.
Chiarella SE, Soberanes S, Urich D, Morales-Nebreda L, Nigdelioglu R, Green D, et al. β2-Adrenergic agonists augment air pollution-induced IL-6 release and thrombosis. J Clin Invest [Internet]. 2014 Jul [cited 2018 Feb 19];124(7):2935–2946. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24865431.
Libalova H, Rossner, P, Vrbova K, Brzicova T, Sikorova J, Vojtisek-Lom M, et al. Comparative Analysis of Toxic Responses of Organic Extracts from Diesel and Selected Alternative Fuels Engine Emissions in Human Lung BEAS-2B Cells. Int J Mol Sci [Internet]. 2016 Nov 3 [cited 2018 mar 8];17(12):1833. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27827897.
Lai C-H, Lee C-N, Bai K-J, Yang Y-L, Chuang K-J, Wu S-M, et al. Protein oxidation and degradation caused by particulate matter. Sci Rep [Internet]. 2016 [cited 2018 Oct 31];6:33727. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27644844.
Surowiec I, Karimpour M, Gouveia-Figueira S, Wu J, Unosson J, Bosson JA, et al. Multi-platform metabolomics assays for human lung lavage fluids in an air pollution exposure study. Anal Bioanal Chem [Internet]. 2016 Jul 25 [cited 2018 Feb 5];408(17):4751–4764. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27113461.
Gouveia-Figueira S, Karimpour M, Bosson JA, Blomberg A, Unosson J, Pourazar J, et al. Mass spectrometry profiling of oxylipins, endocannabinoids, and N-acylethanolamines in human lung lavage fluids reveals responsiveness of prostaglandin E2 and associated lipid metabolites to biodiesel exhaust exposure. Anal Bioanal Chem [Internet]. 2017 Apr 24 [cited 2018 Feb 5];409(11):2967–2980. Available from: http://link.springer.com/10.1007/s00216-017-0243-8
Brown JS. Deposition of Particles. In: Comparative Biology of the Normal Lung [Internet]. Elsevier; 2015 [cited 2018 May 1]. p. 513–536. Available from: http://linkinghub.elsevier.com/retrieve/pii/B9780124045774000278
Kuo C-H, Wang K-C, Tian T-F, Tsai M-H, Chiung Y-M, Hsiech C-M, et al. Metabolomic Characterization of Laborers Exposed to Welding Fumes. Chem Res Toxicol [Internet]. 2012 Mar 19 [cited 2018 Oct 30];25(3):676–686. Available from: http://pubs.acs.org/doi/10.1021/tx200465e
Gu F, Derkach A, Freedman ND, Landi MT, Albanes D, Weinstein SJ, et al. Cigarette smoking behaviour and blood metabolomics. Int J Epidemiol [Internet]. 2016 [cited 2018 Oct 30];45(5):1421–1432. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26721601.
Doberer D, Trejo Bittar HE, Wenzel SE. Should lung biopsies be performed in patients with severe asthma? Eur Respir Rev [Internet]. 2015 Sep [cited 2017 Mar 6];24(137):525–539. Available from: http://err.ersjournals.com/lookup/doi/10.1183/16000617.0045-2015
Kamburov A, Cavill R, Ebbels TMD, Herwig R, Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics [Internet]. 2011 Oct 15;27(20):2917–8 Available from: https://doi.org/10.1093/bioinformatics/btr499.
Koike E, Hirano S, Shimojo N, Kobayashi T. cDNA microarray analysis of gene expression in rat alveolar macrophages in response to organic extract of Diesel exhaust particles. Toxicol Sci [Internet]. 2002 Jun 1;67(2):241–6 Available from: https://doi.org/10.1093/toxsci/67.2.241.
Department for Environment Food and Rural Affairs. Clean Air Strategy 2018 [Internet]. 2018 [cited 2018 Oct 31]. Available from: https://consult.defra.gov.uk/environmental-quality/clean-air-strategy-consultation/user_uploads/clean-air-strategy-2018-consultation.pdf
Lewis AC, Carslaw DC, Kelly FJ. Diesel pollution long under-reported. Nature [Internet]. 2015 Oct 8 [cited 2018 Nov 8];526(7572):195. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26450046.
Acknowledgements
The authors would like to acknowledge Professors Anne Willis and Marion Macfarlane for the kind help and guidance that they provided whilst proof reading this manuscript. Gratitude is also offered to the Medical Research Council for their financial support.
Funding
Production of this manuscript was funded by the Medical Research Council through a UKRI Innovation/ Rutherford Fund Fellowship grant (RG95376) awarded to LS.
Availability of data and materials
Below are links to the author’s original submitted data for cytokine array studies.
Malorni et al. [92]: https://www.sciencedirect.com/science/article/pii/S0378427417300577
Fukagawa et al. [29]: https://pubs.acs.org/doi/abs/10.1021/es403146c
Yanamala et al. [65]: https://www.sciencedirect.com/science/article/pii/S0041008X13003098
Shvedova et al. [90]: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4671493
Author information
Authors and Affiliations
Contributions
IM researched and produced discussion of the health impacts of diesel exhaust while DHP analysed and produced content on studies of biodiesel genotoxicity and mutagenicity. All other sections, including Figs. 1 and 2 and Table 1 were produced by LS. All authors contributed to the editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests, financial or otherwise.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Selley, L., Phillips, D.H. & Mudway, I. The potential of omics approaches to elucidate mechanisms of biodiesel-induced pulmonary toxicity. Part Fibre Toxicol 16, 4 (2019). https://doi.org/10.1186/s12989-018-0284-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12989-018-0284-y