
Abstract
Background
Long non-coding RNAs (lncRNAs) are involved in numerous physiological functions. Yet, their mechanisms in coronary artery disease (CAD) are not well understood.
Methods
The expression profile of genes associated to CAD was reannotated into the lncRNA-mRNA biphasic profile. The target microRNA data were used to design a global CAD triple network. Thereafter, we conducted a functional enrichment analysis and clustering using the triple network from the level of topology analyses. The expression of four non-coding RNAs (ncRNAs) was measured by qRT-PCR and the risk of CAD was calculated by nomogram. The prognostic value of three ncRNAs was evaluated using receiver operating characteristic (ROC) curve.
Results
A CAD lncRNA-miRNA-mRNA network was constructed which included 15 mRNAs, 3 miRNAs, 19 edges and one lncRNA. Nomogram showed that four ncRNAs were the risk of CAD. After RT-PCR validation in four ncRNAs between CAD and non-CAD samples, only three ncRNAs had significant meaning for further analysis. ROC curve showed that TWF1 presented an area under curve (AUC) of 0.862, the AUC of hsa -miR-142-3p was 0.856 and hsa -miR126-5p was 0.822. After the pairwise comparison, we found that TWF1 had significant statistical significance (PTWF1–142 < 0.05 and PTWF1–126 < 0.01). The results of functional enrichment analysis of interacting gene and microRNA showed that the shared lncRNA TWF1 may be a new factor for CAD.
Conclusions
This investigation on the regulatory networks of lncRNA-miRNA-mRNA in CAD suggests that a novel lncRNA, lncRNA TWF1 is a risk factor for CAD, and expands our understanding into the mechanisms involved in the pathogenesis of CAD.
Similar content being viewed by others

Background
Coronary artery disease (CAD) remains one of the most common causes of death worldwide, killing nearly 17 million people each year [1]. In China, it was recently reported that approximately 700,000 deaths from CAD are recorded annually [2]. CAD, as a complex and multifactorial disorder, was caused by lots of environmental exposures and genetic factors, including gender difference, age, dyslipidemia, hypertension, diabetes, obesity, smoking behavior, and family history [3,4,5,6]. Early identification of CAD at high risk and adverse cardiovascular outcomes, using circulating or imaging biomarkers, may help in treatment [7]. However, currently available CAD biomarkers have limited risk prediction [8, 9].
There is increasing evidence that most non-coding RNAs (ncRNAs) have important functions in the modulation of physiological and pathological processes [10, 11]. There are two classes of ncRNA according to the number of nucleotides, including short ncRNAs (< 200 nucleotides) e.g., transcription initiation RNAs, PIWI-interacting RNAs and microRNAs and long lncRNAs (> 200 nucleotides) e.g., enhancer-like ncRNAs and transcribed ultra-conserved regions, natural antisense transcripts and long intergenic ncRNAs (lincRNAs) [12,13,14]. Unlike the highly conserved short ncRNA which inhibits post-transcription, lncRNAs are involved in diverse processes and are not highly conserved [15, 16].
Many studies have shown that many ncRNAs play key roles in specific physiological and pathological processes of CAD [17, 18]. Because of their stability in blood and other body fluids, ncRNAs would be served as biomarkers for cardiovascular diseases (CVDs). Therefore, we hypothesized that there are specific circulating ncRNAs that may serve as CAD biomarkers. Here, a lncRNA-miRNA-mRNA network related to CAD was constructed by mapping the differentially expressed mRNAs, miRNAs and lncRNAs into a global triple network via Gene Expression Omnibus (GEO) repository [19] and want to identify which ncRNAs would be a sensitive and specific marker of CAD.
Materials and methods
Reannotation of gene expression profile probe
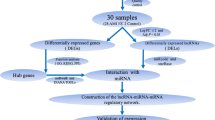
A total of 9 gene expression profile datasets were downloaded from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). The 9 datasets were selected for further bioinformatic analysis, including GSE24519 [20], GSE98895 [21], GSE23561 [22] and GSE56885 for mRNAs; GSE49823, GSE98896 [21] and GSE53675 [23] for miRNAs; and GSE85192 and GSE104815 [24] for lncRNAs. All of the datasets were related to CAD. The transcript sequences of the lncRNAs and proteins were downloaded from the GENCODE database (http://www.gencodegenes.org/). Probe annotation sequence was based on previous reports [25]. Protein coding sequences and probe-matched lncRNA were obtained from Blastn tools. Ideal alignment results were screened based on the following criteria: reserve the probes that: 1) perfectly matched to more than three probes; 2) matched exclusively to a single transcript in probe-transcript pairs; 3) matched to lncRNA transcripts or protein coding transcripts [26]. Affy package in R was used to convert the CEL file to expression value matrix [27], and was quantile normalized using Robust Multi-array Average (RMA) method. Then use the Bioconductor in R to convert the probe information into a gene name [28]. The mean expression value was used for a gene with multiple probes. The specific workflow is shown in Fig. 1.

A flowchart of competing endogenous RNAs (ceRNAs) network construction. (1) DEGs and DELs with fold change ≥2.0 and adjust P-value < 0.05 were used, whereas DEMis was used with fold change ≥1.2 and adjust P-value < 0.05; (2) mRNAs-miRNAs interactions were predicted by miRbase and TargetScan databases; (3) miRNA-lncRNA interactions were predicted by miRcode and starBase databases; (4) lncRNAs which were not associated with DEMis were removed; (5) mRNAs that were not matched DEMis were removed; (6) ceRNA network was constructed
Differentially expressed mRNAs (DEGs), miRNAs (DEMis) and lncRNAs (DELs) and functional enrichment analysis
DEGs, DEMis and DELs were selected using the limma package [29] in R software in all samples. The threshold values for DEGs and DELs were adjusted P < 0.05 and |log2fold-change| ≥ 2, while the threshold values for DEMis were adjusted P < 0.05 and |log2fold-change| ≥ 1.2. Using clusterProfiler, DOSE and Goplot package in R [30, 31], disease ontology (DO), genomes (KEGG) pathway, Kyoto Encyclopedia of Genes and the gene ontology (GO) analyses of DEGs, and GO and DO function analysis of DEMis were carried out. In all of analyses, statistical significance was represented by the adjusted P-value (Q-value) of < 0.05.
Protein-protein interaction analysis (PPI) network construction and module analysis
The STRING database [32] provides information on protein prediction and experimental interactions. The database’s prediction methods come from neighborhoods, databases and text mining, co-expression experiments, co-occurrence and gene fusion. In addition, the interaction of protein pairs in the database was presented as a combined fraction. In this study, DEGs were mapped to PPIs, and a combined score > 0.9 was used as the cutoff value [33, 34], to analyze key genes in the network. The role of protein nodes in the network was described by degrees. Network modules were one of the cores of protein networks and may have specific biological implications. The major clustering modules were examined using the Cytoscape software package Molecular Complex Detection (MCODE) [35, 36] which was used to analyze the most notable clustering modules. Next, the clusterProfiler software package was used to examine the KEGG pathway for DEGs enrichment in different modules. EASE ≤0.05, count ≥2 was set as the cutoff value, and the threshold for the subsequent analysis was MCODE score > 6.
Construction of lncRNA-miRNA-mRNA competing endogenous RNAs (ceRNAs) network
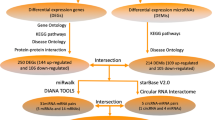
The construction of ceRNA network was derived from the correlation among mRNA, miRNA, and lncRNA. The interactions between lncRNA and miRNA were predicted by the starBase and miRcode databases [37, 38] which were used to predict the lncRNA-miRNA interactions. The target mRNAs of miRNAs were predicted using the miRbase and TargetScan [39, 40]. After GO, DO, KEGG, PPI and MCODE analysis, the DEGs which did not match DEMis should be removed. The same method was used for the lncRNA-miRNA interactions. The lncRNA-miRNA-mRNA ceRNA network was constructed and visualized using the Cytoscape.
Study population
A total of 857 subjects were recruited from outpatient for a complaint of chest in the First Affiliated Hospital, Guangxi Medical University from Jan. 1, 2015 to Dec. 31, 2017. Since angiographic examination was performed for CAD and or other suspected diseases, all participants were examined using coronary angiography by two experts. CAD was defined as significant coronary stenosis (≥ 50%) in at least one of the three main coronary arteries or their major branches (branch diameter ≥ 2 mm). If the coronary stenosis does not meet the above criteria, the subject should be defined as normal coronary artery. To analyze the influence of the expression level of hub gene on the prognosis of the disease, the individuals were divided into CAD and control groups after coronary angiography. Participants who had a history of CAD, type I diabetes mellitus and congenital heart disease were excluded [41]. Medical history and general information were obtained using a standard questionnaire. Thus, the control group was judged to include the subject who with normal coronary artery and the without any excluded disease. The investigation conformed to the rules of the Helsinki Declaration of 1975 (http://www.wma.net/en/30publications/10policies/b3/), and the new edition of 2008. The research design was approved by the Ethics Committee. The First Affiliated Hospital of Guangxi Medical University (No: Lunshen-2011-KY-Guoji-001; March 7, 2011). All procedures are conducted in conformity to ethical standards. All participants singed an informed consent upon receipt of a complete explanation of the study [42]. In the initial evaluation, all clinical data were collected based on the medical records. Clinical data collection, biochemical measurements and diagnostic criteria were performed according to previous studies [43,44,45].
RNA isolation and RT- quantitative PCR (qPCR)
EDTA coated tubes were used to collect fasting blood (5 mL) in the early morning. The blood was centrifuged at 3000 g for 15 min. To remove cell debris, the sample was further centrifuged at 1200 g for 15 min at 4 °C and then kept at − 80 °C. the miRNeasy serum/plasma kit (TIANGEN: Cat. No. DP503, China) was used to isolate total RNA containing miRNA. Briefly, a denaturing buffer was added to 200 μL of plasma sample according to the manufacturer’s instructions. After a 5-min incubation at room temperature, 25 fmol of synthetic cel-miR-39 (TIANGEN; catalog number: CD200–01, China) were added. Thereafter, RNA was isolated using the procedure provided by the manufacturer. RNase-free water (330 μL) was used to elute total RNA. A spectrophotometer (NanoPhotometer P300, Implen) was used to determine the level of hemolysis in plasma samples [46]. cDNA was obtained through a reverse transcription of RNA by a reverse transcriptase kit (TIANGEN; catalog number: KR211, China). The reaction mixture included 10 μL of miRNA RT reaction buffer, 2 μL of enzyme mixture, RNase-free water (up to 20 μL) and 2 μg of total RNA. The mixture was incubated at 42 °C for 60 min, at 95 °C for 3 min, and then at 4 °C. A no RT negative control was used in all tests to ensure that there was no contamination between genomic DNA and PCR product. Quantification of three plasma miRNAs was performed by real-time PCR based on SYBR Green using the miScript SYBR Green PCR Kit (TIANGEN; Cat. No. FP411, China). The reaction contained RNase-free water (up to 20 μL), 3.0 μL cDNA, 0.4 μL PCR reverse primer, 0.2 μL PCR Forward Primer and 2 × miRcute Plus miRNA Premix 10 μL. The reaction was incubated at 95 °C for 15 min, at 94 °C for 25 s, at 60 °C for 30 s, and at 72 °C for 34 s. All reactions were performed in duplicate. Ct values were averaged for each sample after two PCRs experiments. The internal control was for the target RNAs was the cel-miR-39 [47]. The 2 - [Ct (miRNA)- Ct (cel-miR-39)] formula was used to calculate the relative expression of each miRNA after normalization to cel-miR-39. The lncRNA expression levels were quantified in triplicate. The plasma lncRNAs levels were determined by the ∆CT method because there is, as yet, no consensus about stable and suitable internal controls for lncRNA in plasma samples. The change in gene expression was calculated using the equation. 2 −∆CT [48]. The relative expression level of lncRNA in monocytes was normalized to the internal control ACTB expression and calculated by the comparative CT (∆∆CT) method. Melting curve analysis was used to determine the specific and lack of primer dimers for amplification. The primers used in qPCR of the lncRNAs and miRNAs are listed in Additional file 1 Table S1.
Statistical analysis
The statistical software package SPSS 21.0 (SPSS Inc. Chicago, IL, USA) and Prism 5 (GraphPad Software) were used for all statistical analyses. Chi-square analysis was applied to assess differences in ratios among groups. Continuous data are presented as means ± SD [49]. For those that are usually distributed, the median and quartile ranges of triglyceride (TG) are usually not distributed. Comparison of continuous data sets using Mann-Whitney nonparametric tests and Kruskal-Wallis [50]. R software (version 3.5.0) was used to measure the heart-map of correlation models and bioinformatic analysis. The CAD risk score was calculated for each patient as a linear combination of selected predictors that were weighted by their respective coefficients. The ‘rms’ package was used for CAD prediction nomogram. The predictive accuracy of the risk model was assessed by discrimination measured by C-statistic and calibration evaluated by Hosmer-Lemeshow χ2 statistic. To compare the plasma miRNAs and lncRNAs between the control and case groups, receiver operating characteristic (ROC) curve analysis was conducted. The diagnostic value of the miRNAs and lncRNAs was evaluated by the area under curve (AUC). All tests were two-sided, and statistical significance was considered for P < 0.05.
Results
Data preprocessing and identified differentially expressed items
As shown the volcano plot in Fig. 2, After quality control and get rid of lots wrong expression, we identified a total of 752 DEGs including 627 up-regulated and 125 down-regulated in CAD samples compared with control samples with adjust-P value (Pa) < 0.05 and |log 2 (fold change)| ≥ 2; 226 DEMis including 194 up-regulated and 32 down-regulated with Pa < 0.05 and |log 2 (fold change)| ≥ 1.2; 127 DELs including 49 up-regulated and 78 down-regulated with Pa < 0.05 and |log 2 (fold change)| ≥ 2 for further analysis. The detailed items are list in Additional file 1 Tables S1 to S4.

Identified DEGs, DEMis and DELs in volcanoplot. a-d DEGs; (e-g): DEMis; (h-i): DELs. The two vertical lines are the 2-fold change boundaries and the horizontal line is the statistical significance boundary (Adj-P < 0.05). Items with statistical significance and up-regulated are marked with red dots, and down-regulated are marked with green dots
Functional annotation, PPI network construction and identified hub items
To elucidate the role of DEGs, 752 genes were respectively subjected to KEGG pathway enrichment, DO functional and GO analyses by R clusterProfiler package (Fig. 3 a to c). When used the STRING database to analyze, 307 nodes and 1040 protein pairs with a combined weight score > 0.25 were screened in DEGs (Fig. 3d). When they were analyzed in sub-module, only four modules with score > 6 were detected by MCODE (Table 1). The same method was performed to analyze the miRNAs. After GO and DO functional analysis (Fig. 3 e to f), 30 hub miRNAs were identified for further analysis (Table 2).

Functional annotation, PPI network construction and identified hub items. a GO analysis for DEGs; (b): KEGG analysis for DEGs; (c): DO analysis for DEGs. The inner ring is a bar plot where the height of the bar indicates the significance of the term and color corresponds to the z-score. The outer ring displays scatter plots of the expression levels (logFC) for the genes in each term. d PPI network of the selected DEGs. Edge stands for the interaction between two genes. A degree was used for describing the importance of protein nodes in the network, red shows a high degree and white presents a low degree. The significant modules identified from the PPI network using the molecular complex detection method with a score of > 6.0. e GO analysis for DEMis; (f): KEGG analysis for DEMis
Construction of the lncRNA-miRNA-mRNA regulatory network
First of all, considering the interaction between mRNAs and miRNAs, the miRbase and TargetScan databases were searched for miRNA-mRNA interactions. If the predicted genes in the database were not DEGs, they would be removed. At last, we identified 520 miRNA-mRNA interaction pairs (Fig. 4a). We applied the same approach to design the lncRNA-miRNA regulatory network by starBase V2.0 and miRcode and we obtained 8 lncRNA-miRNA interactions pairs. If the predicted miRNAs in the database were not matched DELs, they would also be removed. Finally, a lncRNA-miRNA-mRNA network was formed by merging the two sets of data. LncRNAs, mRNAs and miRNAs were the nodes in the network. The edges in the network represented interactions between these RNAs [51] (Fig. 4b). In total, we found that a lncRNA (TWF1) and three miRNAs (hsa-miR-369-3p, hsa-miR-142-3p and hsa-miR-126-5p) were hub items in this triple regulatory network.

Construction of the lncRNA-miRNA-mRNA regulatory network. a Edge stands for the interaction between two items. Orange shows the up-regulated genes and green presents the down-regulated genes. The significant four modules identified from the PPI network shown with different shapes using the molecular complex detection method with a score of > 6.0. b Edge stands for the interaction between two items. Red shows lncRNAs, orange presents miRNAs and green defines as mRNAs. Different shapes show the different clusters analyzing by the molecular complex detection method
Demographic characteristics, expression levels of ncRNAs and the relationship between ncRNAs and parameters related to CAD
The demographic and biochemical parameters of the participants in the two groups are presented in Table 3. The levels of weight, low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), waist circumference (WC), body mass index (BMI), blood glucose, and the percentage of cigarette smoking and suffered from diabetes were higher in CAD than in control groups. Comparison among age, gender ratio, and the percentage of alcohol drinking revealed no significant difference among groups. As compared with healthy controls, CAD patients had higher circulating TWF1, hsa-miR-126-5p and hsa-miR-142-3p levels (P < 0.05; Fig. 5a). As shown in Fig. 5b, circulating TWF1, hsa-miR-126-5p and hsa-miR-142-3p levels were positively correlated with CAD (P < 0.05 for all).

Confirmed expression levels of ncRNAs and the correlation between ncRNAs and parameters related to CAD. a The relative expression of circulating ncRNAs between healthy controls and CAD samples. b The correlation between ncRNAs and parameters related to CAD. Positive correlation is marked with red, and negative correlation are marked with blue. The colorful lattice with significant difference and leaves blank on no significant coefficient. BMI, Body mass index; SBP, Systolic blood pressure; DBP, Diastolic blood pressure; PP, Pulse pressure; TC, Total cholesterol; TG, Triglyceride; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Apo, Apolipoprotein
Nomogram risk model development to estimate individual CAD probability
We selected gender, age, smoking, drinking, BMI, systolic blood pressure (SBP), diastolic blood pressure (DBP), serum glucose, TC, TG, high-density lipoprotein cholesterol (HDL-C); LDL-C, apolipoprotein (Apo)A1, ApoB, the relative expression of lncRNA TWF1, miR-126-5p, miR-142-3p and miR-369-3p were the best subset of risk factors to develop the CAD risk score and risk model (nomogram) (Fig. 6). Where, male was labeled as 1 and female was labeled as 2; for smoking and drinking, yes was labeled as 2, no was labeled 1. The nomogram had excellent discriminative power with a C-statistic and was well calibrated with Hosmer-Lemeshow χ2 statistic. The predicted probabilities of developing CAD ranging from 0.000054 to 99.9%. After calculation, smoking, drinking serum glucose, ApoB, the relative expression of lncRNA TWF1, miR-126-5p, miR-142-3p and miR-369-3p was significantly related to the risk of CAD, with statistical significance.

Nomogram to estimate individual CAD probability. The nomogram was constructed based on logistic regression model for outcome of definite CAD. Each predictor variable characteristic has a corresponding point value based on its position on the top point scale and contribution to the model. The probability of CAD for each subject is calculated by adding of the points for each variable to obtain a total point value which corresponds to a probability of CAD from the scale presented on the bottom line. *P < 0.05, **P < 0.01
Plasma ncRNAs level sensitive for CAD
As the above-mentioned observations were done, we further assessed these 3 ncRNAs as a marker for CAD. ROC curve analysis showed that TWF1 presented an 95% confidence interval (CI) 0.766–0.958 and AUC of 0.862, the AUC of hsa-miR-142-3p was 0.856 and 95%CI 0.749–0.963 and hsa-miR126-5p was 0.822, 95%CI 0.710–0.933 (Fig. 7a). After the pairwise comparison, we found that TWF1 had significant statistical significance (PTWF1–142 < 0.05; PTWF1–126 < 0.01, Fig. 7b).

ROC curve analyses of these 3 ncRNAs for the diagnosis of CAD. a-c ROC curve analysis of TWF1, hsa-142-3p and hsa-126-5p. d-f The pairwise P-value comparison
Discussion
Novel therapeutic strategies based on mechanisms are required to protect the heart from acute and chronic wound following CVDs. However, the mortality rate remains high. The obvious reason is that CAD is not easy to be found in its earlier stage by regular examinations, such as electrocardiography, cardiac ultrasound. Therefore, it is necessary to detect CAD in its early stage. Emerging evidence has indicated that ncRNAs, such as numerous lncRNAs and miRNAs, modulate many pathophysiological processes in development of CVDs. Indeed, ncRNA in terms of tRNA was identified and its structure was published in the early 1960s [52], accounting for around 97% of the entire genome, while the rest (∼3%) was protein coding genes, since then the research of transcriptomics has expanded and indicated that ncRNAs participated in a wide variety of cellular mechanisms and regulation processes in CVDs [53].
LncRNA belongs to a new class of non-coding RNAs, which are usually defined as transcripts longer than 200 nucleotides and have no protein coding ability [12, 14]. LncRNAs have been implicated in basic biological processes e.g., post-transcriptional gene regulation, gene transcription, chromatin modification, gene regulation and RNA processing [54]. Several lncRNAs are involved in several human diseases e.g., CVD and cancers [55, 56]. For instance, increased lncRNA MEG3 levels may play a critical role in regulating proliferation of endothelial cells and expression of type I collagen, type V collagen resulted in atherosclerosis [57].
MiRNAs play crucial roles in the pathogenesis of CVDs; these miRNAs have also been reported as biomarkers for the prognosis and diagnosis of CAD [58]. Although miRNAs represent a minority of the non-coding transcriptome, the tangle of lncRNAs is likely to contain as yet unidentified classes of molecules, so lncRNAs as diagnostic tools have properties that are advantageous relative to miRNAs. The above-mentioned characteristics make lncRNAs possible to candidate biomarkers for the diagnosis of CAD. The present study compared the lncRNA profiles between the healthy controls and CAD patients and found that plasma lncRNA TWF1 was elevated in the patients with CAD and have an 86.2% specificity for CAD.
Recently, the lncRNA-miRNA-mRNA axes were discovered in CVDs including atherosclerosis, myocardial infarction, and cardiomyopathy, and abnormal conditions such as cardiac fibrosis or/and hypertrophy. For example, the nuclear factor IA (NFIA) regulates cholesterol homeostasis in the body, promoted progression of atherosclerosis through the lncRNA RP5833A20.1 sponging miR-382-5p targeting NFIA axis [59]. These findings suggest that lncRNAs will be candidates for clinical diagnosis and prognostic markers, providing new therapeutic targets for CVDs and providing insights for the prevention and treatment of other diseases in the future. In the current study, we found that several DEGs were associated with CVDs, just as C9orf72 and MBL2 [60, 61], and as the target for hsa-miR-142-3p and hsa-miR-126-5p. Previous research showed that circulating miR126 levels correlated with fasting blood glucose and hemoglobin A1c may lead to CVD [62], and hsa-miR-142-3p up-regulation of miR-142-3p enhances endothelial cell apoptosis and atherosclerotic development which may also contribute to CVD [63]. Two of these miRNAs can be regulatory by TWF1. This result indicates that TWF1 may be a good predictive candidate biomarker for the diagnosis of CAD.
We have to admit the limitation of this study. First, the patients in this study were from one hospital, it is not known whether there is a difference for patients from different areas and races. Therefore, its validity should be tested further in more prospective cohorts. Second, the specific mechanism of lncRNAs-miRNAs-mRNAs axes for regulating the pathogenesis of CAD has not been validated in vivo and in vitro.
Conclusion
Based on the ceRNA hypothesis, construction of lncRNA-miRNA-mRNA triple regulatory network was used to study molecular mechanism of CAD by to accomplish this, we downloaded 9 datasets from GEO and constructed network by combining differentially expressed lncRNAs, miRNAs and mRNAs. After function analysis and predictive ncRNA network construction, we identified regulatory network contained 15 mRNAs, 3 miRNAs, and 1 lncRNA and 19 edges. To further determine the major regulators in this subnetwork, the topological features, function validation and diagnostic prediction were performed which identified one key lncRNA, TWF1.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Apo:
-
Apolipoprotein
- BMI:
-
Body mass index
- CAD:
-
Coronary artery disease
- DBP:
-
Diastolic blood pressure
- DEGs:
-
Differentially expressed mRNAs
- DELs:
-
Differentially expressed lncRNAs
- DEMis:
-
Differentially expressed miRNAs
- DO:
-
Disease ontology
- HDL-C:
-
High-density lipoprotein cholesterol
- LDL-C:
-
Low-density lipoprotein cholesterol
- MCODE:
-
Molecular complex detection
- PP:
-
Pulse pressure
- RMA:
-
Robust Multi-array Average
- ROC:
-
Receiver operating characteristic
- SBP:
-
Systolic blood pressure
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- WC:
-
waist circumference
References
Mortality GBD. Causes of death C: global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the global burden of disease study 2013. Lancet. 2015;385:117–71.
Wang F, Xu CQ, He Q, Cai JP, Li XC, Wang D, Xiong X, Liao YH, Zeng QT, Yang YZ, et al. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat Genet. 2011;43:345–9.
Consortium CAD, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
Souiden Y, Mallouli H, Meskhi S, Chaabouni Y, Rebai A, Cheour F, Mahdouani K. MnSOD and GPx1 polymorphism relationship with coronary heart disease risk and severity. Biol Res. 2016;49:22.
Yamada Y, Matsui K, Takeuchi I, Fujimaki T. Association of genetic variants with coronary artery disease and ischemic stroke in a longitudinal population-based genetic epidemiological study. Biomed Rep. 2015;3:413–9.
Banerjee A, Lim CC, Silver LE, Welch SJ, Banning AP, Rothwell PM. Familial history of stroke is associated with acute coronary syndromes in women. Circ Cardiovasc Genet. 2011;4:9–15.
Malaud E, Merle D, Piquer D, Molina L, Salvetat N, Rubrecht L, Dupaty E, Galea P, Cobo S, Blanc A, et al. Local carotid atherosclerotic plaque proteins for the identification of circulating biomarkers in coronary patients. Atherosclerosis. 2014;233:551–8.
Wykrzykowska JJ, Garcia-Garcia HM, Goedhart D, Zalewski A, Serruys PW. Differential protein biomarker expression and their time-course in patients with a spectrum of stable and unstable coronary syndromes in the integrated biomarker and imaging Study-1 (IBIS-1). Int J Cardiol. 2011;149:10–6.
Eitel I, Blase P, Adams V, Hildebrand L, Desch S, Schuler G, Thiele H. Growth-differentiation factor 15 as predictor of mortality in acute reperfused ST-elevation myocardial infarction: insights from cardiovascular magnetic resonance. Heart. 2011;97:632–40.
Aryal B, Rotllan N, Fernandez-Hernando C. Noncoding RNAs and atherosclerosis. Curr Atheroscler Rep. 2014;16:407.
Fu XD. Non-coding RNA: a new frontier in regulatory biology. Natl Sci Rev. 2014;1:190–204.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Dechamethakun S, Muramatsu M. Long noncoding RNA variations in cardiometabolic diseases. J Hum Genet. 2017;62:97–104.
Elia L, Condorelli G. RNA (epi)genetics in cardiovascular diseases. J Mol Cell Cardiol. 2015;89:11–6.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62.
Schonrock N, Harvey RP, Mattick JS. Long noncoding RNAs in cardiac development and pathophysiology. Circ Res. 2012;111:1349–62.
Mathiyalagan P, Keating ST, Du XJ, El-Osta A. Interplay of chromatin modifications and non-coding RNAs in the heart. Epigenetics. 2014;9:101–12.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991–5.
Su J, Gao C, Wang R, Xiao C, Yang M. Genes associated with inflammation and the cell cycle may serve as biomarkers for the diagnosis and prognosis of acute myocardial infarction in a Chinese population. Mol Med Rep. 2018;18:1311–22.
D'Amore S, Hardfeldt J, Cariello M, Graziano G, Copetti M, Di Tullio G, Piglionica M, Scialpi N, Sabba C, Palasciano G, et al. Identification of miR-9-5p as direct regulator of ABCA1 and HDL-driven reverse cholesterol transport in circulating CD14+ cells of patients with metabolic syndrome. Cardiovasc Res. 2018;114:1154–64.
Grayson BL, Wang L, Aune TM. Peripheral blood gene expression profiles in metabolic syndrome, coronary artery disease and type 2 diabetes. Genes Immun. 2011;12:341–51.
Xu Z, Han Y, Liu J, Jiang F, Hu H, Wang Y, Liu Q, Gong Y, Li X. MiR-135b-5p and MiR-499a-3p promote cell proliferation and migration in atherosclerosis by directly targeting MEF2C. Sci Rep. 2015;5:12276.
Liu Y, Ji Y, Li M, Wang M, Yi X, Yin C, Wang S, Zhang M, Zhao Z, Xiao Y. Integrated analysis of long noncoding RNA and mRNA expression profile in children with obesity by microarray analysis. Sci Rep. 2018;8:8750.
Zhang X, Sun S, Pu JK, Tsang AC, Lee D, Man VO, Lui WM, Wong ST, Leung GK. Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiol Dis. 2012;48:1–8.
Song C, Zhang J, Liu Y, Pan H, Qi HP, Cao YG, Zhao JM, Li S, Guo J, Sun HL, Li CQ. Construction and analysis of cardiac hypertrophy-associated lncRNA-mRNA network based on competitive endogenous RNA reveal functional lncRNAs in cardiac hypertrophy. Oncotarget. 2016;7:10827–40.
Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–15.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80.
Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004;20:3705–6.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–4.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52.
Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–2.
Zhang H, Zhang X, Huang J, Fan X. Identification of key genes and pathways for peri-implantitis through the analysis of gene expression data. Exp Ther Med. 2017;13:1832–40.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4(2).
Jeggari A, Marks DS, Larsson E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics. 2012;28:2062–3.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–7.
Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73.
Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4.
Miao L, Yin RX, Yang S, Huang F, Chen WX, Cao XL. Association between single nucleotide polymorphism rs9534275 and the risk of coronary artery disease and ischemic stroke. Lipids Health Dis. 2017;16:193.
Miao L, Yin RX, Huang F, Chen WX, Cao XL, Wu JZ. The effect of MVK-MMAB variants, their haplotypes and GxE interactions on serum lipid levels and the risk of coronary heart disease and ischemic stroke. Oncotarget. 2017;8:72801–17.
Miao L, Yin RX, Wu JZ, Yang S, Lin WX, Pan SL. The SRGAP2 SNPs, their haplotypes and G x E interactions on serum lipid traits. Sci Rep. 2017;7:11626.
Miao L, Yin RX, Pan SL, Yang S, Yang DZ, Lin WX. BCL3-PVRL2-TOMM40 SNPs, gene-gene and gene-environment interactions on dyslipidemia. Sci Rep. 2018;8:6189.
Miao L, Yin RX, Pan SL, Yang S, Yang DZ, Lin WX. Weighted gene co-expression network analysis identifies specific modules and hub genes related to hyperlipidemia. Cell Physiol Biochem. 2018;48:1151–63.
Quan X, Ji Y, Zhang C, Guo X, Zhang Y, Jia S, Ma W, Fan Y, Wang C. Circulating MiR-146a may be a potential biomarker of coronary heart disease in patients with subclinical hypothyroidism. Cell Physiol Biochem. 2018;45:226–36.
Aroldi A, Lampertico P, Montagnino G, Passerini P, Villa M, Campise MR, Lunghi G, Tarantino A, Cesana BM, Messa P, Ponticelli C. Natural history of hepatitis B and C in renal allograft recipients. Transplantation. 2005;79:1132–6.
Ren S, Wang F, Shen J, Sun Y, Xu W, Lu J, Wei M, Xu C, Wu C, Zhang Z, et al. Long non-coding RNA metastasis associated in lung adenocarcinoma transcript 1 derived miniRNA as a novel plasma-based biomarker for diagnosing prostate cancer. Eur J Cancer. 2013;49:2949–59.
Zhang QH, Yin RX, Huang F, Yang DZ, Lin WX, Pan SL. Association between the PINX1 and NAT2 polymorphisms and serum lipid levels. Oncotarget. 2017;8:114081–94.
Miao L, Yin RX, Pan SL, Yang S, Yang DZ, Lin WX. Association between the MVK and MMAB polymorphisms and serum lipid levels. Oncotarget. 2017;8:70378–93.
Wang P, Li J, Zhao W, Shang C, Jiang X, Wang Y, Zhou B, Bao F, Qiao H. A novel LncRNA-miRNA-mRNA triple network identifies LncRNA RP11-363E7.4 as an important regulator of miRNA and gene expression in gastric Cancer. Cell Physiol Biochem. 2018;47:1025–41.
He L, Chen Y, Hao S, Qian J. Uncovering novel landscape of cardiovascular diseases and therapeutic targets for cardioprotection via long noncoding RNA-miRNA-mRNA axes. Epigenomics. 2018;10:661–71.
Boon RA, Jae N, Holdt L, Dimmeler S. Long Noncoding RNAs from Clinical Genetics to Therapeutic Targets? J Am Coll Cardiol. 2016;67:1214–26.
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58.
Spizzo R, Almeida MI, Colombatti A, Calin GA. Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene. 2012;31:4577–87.
Jaipersad AS, Shantsila A, Lip GY, Shantsila E. Expression of monocyte subsets and angiogenic markers in relation to carotid plaque neovascularization in patients with pre-existing coronary artery disease and carotid stenosis. Ann Med. 2014;46:530–8.
Wu Z, He Y, Li D, Fang X, Shang T, Zhang H, Zheng X. Long noncoding RNA MEG3 suppressed endothelial cell proliferation and migration through regulating miR-21. Am J Transl Res. 2017;9:3326–35.
Wang K, Liu CY, Zhou LY, Wang JX, Wang M, Zhao B, Zhao WK, Xu SJ, Fan LH, Zhang XJ, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015;6:6779.
Hu YW, Zhao JY, Li SF, Huang JL, Qiu YR, Ma X, Wu SG, Chen ZP, Hu YR, Yang JY, et al. RP5-833A20.1/miR-382-5p/NFIA-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis and inflammatory reaction. Arterioscler Thromb Vasc Biol. 2015;35:87–101.
Smith NL, Felix JF, Morrison AC, Demissie S, Glazer NL, Loehr LR, Cupples LA, Dehghan A, Lumley T, Rosamond WD, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–66.
van der Harst P, van Setten J, Verweij N, Vogler G, Franke L, Maurano MT, Wang X, Mateo Leach I, Eijgelsheim M, Sotoodehnia N, et al. 52 genetic loci influencing myocardial mass. J Am Coll Cardiol. 2016;68:1435–48.
Park S, Moon S, Lee K, Park IB, Lee DH, Nam S. Urinary and blood MicroRNA-126 and -770 are potential noninvasive biomarker candidates for diabetic nephropathy: a meta-analysis. Cell Physiol Biochem. 2018;46:1331–40.
Qin B, Shu Y, Long L, Li H, Men X, Feng L, Yang H, Lu Z. MicroRNA-142-3p induces atherosclerosis-associated endothelial cell apoptosis by directly targeting Rictor. Cell Physiol Biochem. 2018;47:1589–603.
Acknowledgments
We thank all the staff in the laboratory.
Funding
The authors acknowledge the essential role of the funding of the National Natural Science Foundation of China (No: 81460169) and the Innovation Project of Guangxi Graduate Education in this motif.
Author information
Authors and Affiliations
Contributions
L.M. conceived the study, participated in the design, undertook genotyping, performed the statistical analyses, and drafted the manuscript. R.-X.Y. conceived the study, participated in the design, carried out the epidemiological survey, collected the samples, and helped to draft the manuscript. Q.-H.Z. H.-X.J. and F.H. collaborated to the genotyping. W.-X.C., X.-L.C. and J.-Z.W. carried out the epidemiological survey and collected the samples. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research design was approved by the Ethics Committee. The First Affiliated Hospital of Guangxi Medical University (No: Lunshen-2011-KY-Guoji-001; March 7, 2011). All procedures are conducted in conformity to ethical standards.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. PCR primers for quantitative real-time PCR. Table S2. Differentially expressed mRNAs (DEGs). Table S3. Differentially expressed miRNAs (DEMis). Table S4. Differentially expressed lncRNAs (DELs). (XLSX 113 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Miao, L., Yin, RX., Zhang, QH. et al. A novel lncRNA-miRNA-mRNA triple network identifies lncRNA TWF1 as an important regulator of miRNA and gene expression in coronary artery disease. Nutr Metab (Lond) 16, 39 (2019). https://doi.org/10.1186/s12986-019-0366-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12986-019-0366-3