
Abstract
Background
The chemokine receptor CCR5, which belongs to the superfamily of G protein-coupled receptors, is the major co-receptor for HIV-1 entry. Individuals with a homozygous CCR5Δ32 mutation have a long lasting and increased resistance to HIV-1 infection. Therefore, CCR5 represents an optimal target for HIV-1/AIDS gene therapy. The CRISPR/Cas9 system has been developed as one of the most efficacious gene editing tools in mammalian cells and the small-sized version from Staphylococcus aureus (SaCas9) has an advantage of easier delivery compared to the most commonly used version from Streptococcus pyogenes Cas9 (SpCas9).
Results
Here, we demonstrated that CCR5 could be specifically and efficiently edited by CRISPR/SaCas9 together with two sgRNAs, which were identified through a screening of 13 sgRNAs. Disruption of CCR5 expression by lentiviral vector-mediated CRISPR/SaCas9 led to increased resistance against HIV-1 infection in human primary CD4+ T cells. Moreover, humanized mice engrafted with CCR5-disrupted CD4+ T cells showed selective survival and enrichment when challenged with CCR5 (R5)-tropic HIV-1 in comparison to mock-treated CD4+ T cells. We also observed CCR5 could be targeted by CRISPR/SaCas9 in human CD34+ hematopoietic stem/progenitor cells without obvious differentiation deficiencies.
Conclusions
This work provides an alternative approach to disrupt human CCR5 by CRISPR/SaCas9 for a potential gene therapy strategy against HIV-1/AIDS.
Similar content being viewed by others


Background
Although the clinical application of highly active antiretroviral therapy (HAART) effectively inhibits HIV-1 replication and prolongs lifespan of the patients with acquired immunodeficiency syndrome (AIDS), it cannot eradicate the latent reservoir of the virus [1, 2]. Additionally, HAART has limitations due to its high cost, drug resistance, requirement for long-term adherence to treatment, and side effects such as toxicity and even immune dysfunction [3,4,5,6,7]. Therefore, it is necessary to look for more effective approaches to eliminate HIV-1 proviral DNA in the latent reservoir of infected individuals and pursue a cure for HIV-1/AIDS patients. In recent decades, gene therapy has been developed as a new strategy for improving the health of patients with genetic diseases, such as hemophilia [8], β-thalassemia [9] and other monogenic diseases [10]. The strategies involve using nucleases for specific gene editing to cure disease. In previous studies, zinc finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) were the two special nucleases which could recognize the genomic editing locus by protein–DNA interaction [11, 12] and both of them had been applied in generating resistance to HIV-1/AIDS infection [13,14,15]. However, these two strategies have several limitations in application, including low editing efficiency and time-consuming production. In recent years, clustered regularly interspaced short palindromic repeats (CRISPR) in complex with CRISPR-associated protein 9 (Cas9) has been widely used for gene editing in mammalian cells. The Cas9 proteins derived from Streptococcus pyogenes (SpCas9) or Staphylococcus aureus (SaCas9), combined with a single small guide RNA (sgRNA) and type II CRISPR system from bacteria, can recognize and cleave DNA loci followed by a 5′-protospacer adjacent motif (PAM) sequence of NGG and NNGRRT, respectively [16,17,18,19]. DNA cleavage induces double-stranded DNA breaks (DSBs), which are repaired via error-prone non-homologous end joining (NHEJ) or homologous recombination (HR) in eukaryotes, resulting in deletions and insertions (indels) or substitution in the target sequences of the genome [19].
HIV-1 enters into cells via initial binding of gp120 envelope protein to the cellular receptor CD4 [20], followed by one of the two chemokine co-receptor CCR5 or CXCR4 [21, 22]. CCR5 is the major co-receptor for CCR5 (R5)-tropic HIV-1 [23], while CXCR4 is used as the co-receptor for CXCR4 (X4)-tropic HIV-1 that appears in about half of late-stage infections [24]. Previous studies showed that individuals with the naturally occurring CCR5Δ32 mutation were resistant to HIV-1 infection [25, 26]. Further, the ‘Berlin patient’, an individual with acute myelocytic leukemia (AML) and HIV-1/AIDS, lived free of HIV-1 infection after receiving bone marrow from a donor with the CCR5Δ32 genotype, suggesting a key role for CCR5 in HIV-1 infection [27, 28]. In addition, a recent report about the ‘London patient’ with Hodgkin’s lymphoma provides evidence for HIV-1 remission by CCR5Δ32 hematopoietic stem-cell (HSC) transplantation [29]. Thus, it is important to develop HIV cure strategies based on preventing or disrupting the expression of CCR5 co-receptor. Previous reports suggested that specific targeting of CCR5 in human autologous CD4+ T cells by ZFN, TALEN or CRISPR/SpCas9 protected against HIV-1 infection [13, 15, 30,31,32]. Additionally, efficient ablation of CCR5 had been achieved in human hematopoietic stem/progenitor cells and induced pluripotent stem cells by CRISPR/SpCas9 [33,34,35,36]. In recent years, a smaller SaCas9 has attracted more attention for its effective gene editing ability and ease of delivery. The adeno-associated virus (AAV)-SaCas9 system has been successfully applied in gene knock-in and knock-out studies, suggesting the possibility for SaCas9 used in HIV-1/AIDS gene therapy researches [18, 37,38,39,40]. Indeed, previous researches had shown that disruption of co-receptor CXCR4 and HIV-1 provirus by SaCas9/gRNAs promoted human primary CD4+ T cells and Jurkat T cells resistance to HIV-1 infection [41, 42]. It had also been reported that excision of HIV-1 provirus by SaCas9 and multiplex sgRNAs had been achieved in humanized mice models [40]. Therefore, the CRISPR/SaCas9 system is considered as a beneficial and effective gene editing tool with potential to be an HIV-1/AIDS treatment strategy.
In this study, we identified two sgRNAs that could guide SaCas9 specifically and efficiently to target CCR5. By using a lentiviral vector for delivery, we observed efficient editing of CCR5 in primary human CD4+ T cells, leading to cell resistance to HIV-1 infection. Moreover, we showed survival and enrichment of CCR5-disrupted CD4+ T cells in humanized mice during R5-tropic HIV-1 infection. We also demonstrated that SaCas9/sgRNA induced CCR5 editing in CD34+ hematopoietic stem/progenitor cells without obvious differentiation deficiencies. Together, our data suggest that CCR5 can be effectively edited by CRISPR/SaCas9 with selected target sgRNAs and a small-sized SaCas9, which may provide an alternative approach for CCR5 disruption in HIV-1/AIDS gene therapy.
Results
RNA-guided SaCas9 nuclease mediates efficient disruption of CCR5 to protect TZM-bl cells from R5-tropic HIV-1 infection
To identify effective target sites, we used an online tool (http://crispr.cos.uni-heidelberg.de/) to design 13 sgRNAs with the PAM sequence of 5′-NNGRRT-3′ to target the open reading frame (ORF) of CCR5, in addition, we used an effective sgRNA of CXCR4 [41] as a negative control for targeting of CCR5 (Additional file 1: Fig. S1; Additional file 4: Table S1). To select efficient sgRNAs, we first inserted all designed target DNA (referred to as sgRNAs) into an AAV-CRISPR-SaCas9 (PX601) plasmid (Fig. 1a). We then tested the efficiency of the AAV-CRISPR/SaCa9 system and targeting of the CCR5 gene by the sgRNAs in HeLa cells. Three days after transfection of AAV-SaCas9/sgRNA, we conducted T7 endonuclease 1 (T7E1) assays, which could detect and cleave mismatched DNA. The result showed that the 1054 bp PCR products from the cell genome could be edited by sgRNA-#2, #6, # 8 and #11 delivered by AAV-CRISPR-SaCas9, while the negative sgRNA and control showed no cleavage (Additional file 2: Fig. S2). We also found that sgRNA-#6 and #8 had much higher gene editing efficiency than others (Additional file 2: Fig. S2).

Efficient disruption of CCR5 in TZM-b1 cells by AAV-CRISPR/SaCas9 defenses HIV-1 infection. a The diagram of AAV-CRISPR-SaCas9 and Lenti-CRISPR-SaCas9 vector composition and insert sites of CCR5-sgRNA. b T7E1 assay analysis of disruption efficiency in TZM-bl cells transfected with AAV-CRISPR/SaCas9-sgRNA-#6, #8 or control. The indel percentage was calculated using Image J software. c Flow cytometry detection of CCR5 expression on cell surface. The TZM-bl cells transfected with AAV-SaCas9/sgRNA-#6, #8 or control were stained with APC-conjugated CCR5 antibody and analyzed by flow cytometry. The unstained control-transfected TZM-bl cells and stained control-transfected TZM-bl cells were treated as negative and positive controls respectively. The data showed on the top of each peak were the percentage of CCR5 negative and positive cells. d DNA sequences of CCR5 target sites in the TZM-bl cells mediated by sgRNA-#6 and #8. PCR products amplification from genomic DNA were cloned into T-vector and performed Sanger sequencing. The red sequences indicate the PAM sequences; the blue sequences are marked as the targeted sequences; the green bases in the targeted sequences are mutations; insertions and deletions are indicated with (+) and (−) respectively. N/N shows the ratio of mutations or wild type (WT) in the all sequenced clones. e Disruption of CCR5 in TZM-bl cells resistance to HIV-1 infection. TZM-bl cells edited by sgRNA-#6, #8 or control were infected with R5-tropic HIV-1YU2 (MOI = 0.5) and analyzed by luciferase reporter assay. Data were analyzed by unpaired t-test and error bars showed the mean ± SEM of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
To further confirm the editing efficiency of sgRNA-#6 and #8 in an HIV-1 reporter cell line, we then transfected AAV-SaCas9/sgRNAs or control into the TZM-bl cells, an HIV-1-susceptible cell line originally adapted from HeLa cells expressing human CD4, CCR5, and an HIV-1 LTR-driven luciferase reporter [43, 44]. We conducted a T7E1 assay 3 days post-transfection. Our results showed that PCR products of the CCR5 gene in TZM-bl cells could be cleaved into two fragments by both sgRNAs, suggesting that sgRNA-#6 and #8 efficiently induced CCR5 disruption in TZM-bl cells (Fig. 1b). To further analyze whether the sgRNAs could disrupt CCR5 expression in TZM-bl cells, we measured the protein levels of CCR5 on the cell surface 3 days post-transfection by flow cytometry. Results indicated a 10.6% and 21.5% reduction of CCR5 expression upon treatment with sgRNA-#6 and #8, respectively (Fig. 1c). We then inserted the two PCR products of the target loci into a T-vector and determined the indels by DNA sequencing. The results showed that sgRNA-#6 and #8 induced indels and mutations in CCR5 gene (Fig. 1d). Next, we determined whether the disruption of CCR5 expression by selected sgRNA-guided SaCas9 cleavage could resist HIV-1 infection. We transfected TZM-bl cells with AAV-SaCas9/sgRNA-#6, #8 or control and then infected the modified cells with R5-tropic HIV-1YU2 strain. Three days post-infection, the cells were collected and a luciferase reporter assay was performed to analyze the infection efficiency. We observed that HIV-1 infection levels in CCR5-edited cells were significantly reduced compared to the control-treated cells (Fig. 1e).
As sgRNA-#6 and sgRNA-#8 functioned efficiently in the CRISPR/SaCas9 system delivered by AAV vector, we wanted to know whether they could be delivered by lentiviral vector. In order to construct the Lenti-CRISPR/SaCas9 system, we replaced SpCas9 in the lentiCRISPR-v2 plasmid with the PCR-amplified SaCas9 from the PX601 plasmid. DNA targets of sgRNA-#6 and sgRNA-#8 were then inserted into the Lenti-CRISPR/SaCas9 recombinant vector (Fig. 1a). After analyzing the system by sequencing, we transfected Lenti-SaCas9/sgRNA-#6, #8 or control into TZM-bl cells. The T7E1 assay of the cell genome indicated that PCR products of CCR5 gene could be cleaved by sgRNA-#6 and sgRNA-#8 efficiently (Fig. 2a). Further, flow cytometry analysis demonstrated that the reduction of CCR5 expression mediated by sgRNA-#6 and #8 could reach 45.5% and 58.8% respectively compared to control (Fig. 2b). Lastly, results of DNA sequencing (Fig. 2c) and the HIV-1 challenge assay (Fig. 2d) showed that Lenti-SaCas9/sgRNA-#6 and #8 could achieve highly efficient disruption of CCR5 and thus confer cell resistance against HIV-1 infection. All together, these results demonstrated that disruption of CCR5 in HIV-1 reporter cell lines by SaCas9/sgRNA-#6 and #8 protected cells from R5-tropic HIV-1 infection and sgRNAs delivered by Lenti-CRISPR/SaCas9 had higher efficiency than AAV- CRISPR/SaCas9.

LentiCRISPR/SaCas9 mediated cleavage of CCR5 in TZM-bl cells against HIV-1 infection. a LentiCRISPR/SaCas9 editing of the CCR5 gene in TZM-bl cells. sgRNA-#6, #8 or control were transfected into TZM-bl cells delivered by lentiviral vector. Three days post transduction, genomic DNA was extracted and T7E1 assay was conducted. b Detection of CCR5 expression in TZM-bl cells by flow cytometry. c Sanger sequencing of CCR5 target loci in the TZM-bl cells modified by LentiCRISPR-SaCas9/sgRNA. d Disruption of CCR5 in TZM-bl cells could render cell resistance to HIV-1 infection. The cells were transduced with LentiCRISPR-SaCas9/sgRNA-#6, #8 or control. Then the modified cells were infected with HIV-1YU2 (MOI = 0.5). Luciferase reporter assay was performed to analyze infection efficiency. Data were analyzed by unpaired t-test and error bars showed the mean ± SEM of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
Disruption of CCR5 in Jurkat T cells by LentiCRISPR/SaCas9 confers resistance to R5-tropic HIV-1 infection
To recapitulate the CCR5 gene editing results obtained in TZM-bl cells, we tested whether sgRNA-#6 and #8 together with SaCas9 could disrupt CCR5 expression in a CD4+ T cell line. After several unsuccessful attempts to disrupt CCR5 in Jurkat T cells by AAV-CRISPR/SaCas9, we conducted lentiviral vector-mediated transduction of SaCas9/sgRNA-#6, #8 or control into Jurkat T cells to disrupt the CCR5 gene. Three days after transduction, T7E1 assay was performed to determine the disruption efficiency of CCR5. The result showed that sgRNA-#6 and #8 could induce CCR5 mutation in Jurkat T cells with high efficiency (Fig. 3a). Consistently, immunoblotting of the transduced cells indicated that the expression of CCR5 on the surface of Jurkat T cells was markedly reduced upon treatment with sgRNA-#6 and #8 compared to control (Fig. 3b). In a parallel assay, DNA sequencing showed indel mutations in the CCR5 targeting sites (Fig. 3c), suggesting that the CCR5 gene in Jurkat T cells could be efficiently edited by lentiviral-mediated SaCas9/sgRNA delivery.

Disruption of CCR5 in Jurkat T cells protects cells from HIV-1 infection via LentiCRISPR/SaCas9. a Mutation of CCR5 in Jurkat T cells was detected by T7E1 assay. Jurkat T cells were transduced with SaCas9/sgRNA-#6, #8 or control lentivirus with MOI of 40. After 3 days selection by puromycin, the cells were harvested for T7E1 assay. b Western blotting assessment of CCR5 expression on SaCas9/sgRNA-lentivirus edited Jurkat T cells. CCR5-modified or control Jurkat T cells in a were collected for western blotting assay with anti-CCR5 and anti-GAPDH antibodies. c DNA sequencing of CCR5 gene target loci in the modified Jurkat T cells. d The CCR5-sgRNA transduced Jurkat T cells attenuated HIV-1YU2 infection. The SaCas9/sgRNA-#6, #8 or control-treated Jurkat T cells were infected with R5-tropic HIV-1YU2 (MOI = 0.1). After 1, 3, 5, 7 days infection, cell culture medium was collected for p24 ELISA assay. Data were analyzed by unpaired t-test and error bars showed the mean ± SEM of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
To determine whether disruption of CCR5 in Jurkat T cells by Lenti-SaCas9/sgRNA could result in increased resistance to HIV-1 infection, Jurkat T cells with CCR5 mutation induced by sgRNA-#6, #8 or control were challenged with R5-tropic HIV-1YU2. At 1, 3, 5, and 7 days post-infection, we collected cellular supernatant and performed an ELISA assay for HIV-1 p24. The results indicated that the level of p24 in CCR5-edited Jurkat T cells decreased significantly compared to that in control (Fig. 3d). These results demonstrated that sgRNA-#6 and #8 delivered by lentiviral vector could efficiently induce SaCas9 cleavage of the CCR5 gene in Jurkat T cells, leading to increased cell resistance to HIV-1 infection.
LentiCRISPR/SaCas9-mediated CCR5 gene editing in human primary CD4+ T cells confers resistance to R5-tropic HIV-1 infection
After successfully disrupting CCR5 in both an HIV-1 report cell line and a CD4+ T cell line, we next attempted to deliver LentiCRISPR/SaCa9 combined with sgRNA-#6, #8 or control plasmid into human primary CD4+ T cells to disrupt CCR5 expression. Human primary CD4+ T cells isolated from healthy donors were transduced with packaged lentivirus with an MOI of 100. Three days post-transduction, we performed the T7E1 assay and found that both sgRNA-#6 and #8 could induce CCR5 editing in primary human CD4+ T cells (Fig. 4a). However, the gene editing efficiency was lower than that in Jurkat T cells, which may be due to the low lentiviral transduction efficiency in primary CD4+ T cells [31]. Immunoblotting showed that the expression of CCR5 decreased in sgRNA-#6 and #8 modified CD4+ T cells compared with control (Fig. 4b). To further confirm the disruption of CCR5 by SaCas9/sgRNA-#6 and #8 in human CD4+ T cells, we sequenced 10 random PCR products in the T-vector and found indels and mutations in the target region (Fig. 4c). To evaluate whether the modification of CCR5 in primary human CD4+ T cells by CRISPR/SaCas9 could render the cells resistant to HIV-1 infection, we infected the CD4+ T cells with HIV-1YU2 and then cultured these cells for 7 days. The assessment of virus by measuring the HIV-1 p24 in cell culture medium at days 1, 3, 5, and 7 post-infection, which demonstrated a decrease in the p24 level over time compared to control (Fig. 4d). These data indicated that CCR5-edited in human primary CD4+ T cells by LentiCRISPR/SaCas9 could inhibit R5-tropic HIV-1 infection.

Ablation of CCR5 by LentiCRISPR/SaCas9 in primary human CD4+ T cells confers HIV-1 resistance. a Disruption of CCR5 in primary CD4+ T cells by LentiCRISPR/SaCas9. The primary CD4+ T cells were transduced with LentiCRISPR-SaCas9/sgRNA-sgRNA-#6, #8 or control with MOI of 100. After 72 h transduction, cells were harvested for T7E1 assay. b Detection of CCR5 expression in primary CD4+ T cells modified by LentiCRISPR/SaCas9-sgRNA-#6. #8 or control. Modified CD4+ T cells were lysed and analyzed by western blotting with anti-CCR5 and anti-GAPDH antibodies. c Sanger sequencing of CCR5 gene target loci in modified primary CD4+ T cells. d Ablation of CCR5 in CD4+ T cells resistance to HIV-1 infection. The CD4+ T cells edited by SaCas9/sgRNA-#6, #8 or control were treated with HIV-1YU2 (MOI = 0.1) for 8 h and then cell culture medium was changed with fresh medium. The supernatant was collected at the setting time points for p24 ELISA assay. Data were analyzed by unpaired t-test and error bars showed the mean ± SEM of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
CCR5-modified human primary CD4+ T cells have survival advantages after challenge with HIV-1 in humanized mice
Immunodeficient mice are commonly used to model HIV-1 infection in vivo [33, 34, 45]. As we demonstrated the efficient disruption of CCR5 by SaCas9/sgRNA in vitro, we wanted to evaluate whether CRISPR/SaCas9-mediated disruption of CCR5 could protect primary CD4+ T cells against HIV-1 infection in vivo. We chose the most efficient sgRNA (#8) and injected two groups of NOD-Prkdcem26Cd52Il2rgem26Cd22/Nju (NCG) humanized mice with mock-modified human CD4+ T cells or LentiCRISPR-SaCas9/sgRNA-#8-modified CD4+ T cells (n = 8 per group) (Fig. 5a). The human CD4+ T cell counts in the peripheral blood of each mouse were evaluated by flow cytometry 28 days post-injection to assess engraftment. 7 days after assessing engraftment, we injected and infected half of the mice (n = 4) in each group with or without HIV-1YU2 (Fig. 5a).

CCR5 disruption via LentiCRISPR/SaCas9 renders CD4+ T cells survival from HIV-1 infection in vivo. a Scheme of humanized mice experiment. Experiment schedule and process were showed at each time point. 16 mice were divided into two groups randomly and each group received mock-modified or SaCas9/sgRNA-#8-CCR5-modified human CD4+ T cells. b Assess engraftment of all the mice post injection by flow cytometry to calculate CD4+ T cell counts. c Analysis of CD4+ T cell counts after infection with HIV-1. CD4+ T cells counts were calculated at 14, 28, 45 days post-infection by flow cytometry. d Measure the proportion of human CD4+ T cells in spleens of HIV-1 infected mice after euthanasia. e The relative copy numbers of gag gene in CD4+ T cells of spleens in d. f CCR5 alleles mutation in CD4+ T cells from humanized mice. The human CD4+ T lymphocytes from spleens were conducted for genomic DNA extraction and T7E1 assay after 45 days infection. Mutation were showed in CCR5-modified CD4+ T cells in mice. Data were analyzed by unpaired t-test. Error bars showed the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001)
We monitored the counts of CD4+ T cells in each group at different time points to assess the effect of HIV-1 infection in cells modified with CRISPR/SaCas9-sgRNA-#8 in vivo (Fig. 5a). Before HIV-1 infection, the cell counts in the group with mock-modified cells were higher than that with R5-modified cells (Fig. 5b). This slight engraftment difference may be donor-specific, as shown in a previous study [34]. However, in the R5-HIV-1 infection group, the difference in cell counts between mock-modified and R5-modified group became more obvious with time (Fig. 5c). 14 days post-infection, CCR5-modified cells in R5-HIV-1 infected group were ~ten-fold higher in number than mock-modified cells in R5-HIV-1 infected group (Fig. 5c, left panel). 28 days post-infection, however, we did not observe marked differences in CD4+ T cell counts with HIV-1 infection in mock group compared to the R5-modified group (Fig. 5c, middle panel), which may be due to the killing of unmodified CD4+ T cell by HIV-1 infection in R5-modified group. 45 days post-infection, CD4+ T cell counts of the R5-modified, HIV-1-infected group were about eight-fold higher than that of the mock-modified, HIV-1-infected group, and the difference was statistically significant (Fig. 5c, right panel). As expected, the majority of mice injected with R5-modified or mock-modified CD4+ T cells showed xenogeneic graft versus host disease (XGVHD) with clinical features of hair loss and dermatitis, which often appear between 47 and 52 days post-engraftment [46]. We sacrificed the mice that had developed XGVHD and the final numbers of the mice that remained were equivalent in modified and unmodified groups, indicating that R5-SaCas9 itself might not alter CD4+ T cell function and have few side effects in vivo.
Furthermore, we determined the percentage of human CD4+ T cells in the spleen after euthanasia of the HIV-infected mice. The mice that received R5-modified CD4+ T cells had about 4% human CD4+ T cells of total cells in spleens. In contrast, human CD4+ T cells were almost undetectable in the spleens of the mice infused with mock-modified CD4+ T cells (Fig. 5d). We also performed real-time PCR assay to detect relative copy number of the HIV-1 gag gene in human CD4+ T cells in the spleens, and the results demonstrated a lower copy number of gag normalized to β-globin in R5-modified CD4+ T cells infected with HIV-1 compared to that of control (Fig. 5e). Importantly, T7E1 assay of the CCR5 gene in CD4+ T cells from the spleens showed the mutation in CCR5-modified CD4+ T cells with or without HIV-1 infection (Fig. 5f), which was indicative of successful modification and engraftment of CD4+ T cells in humanized mice. In addition, R5-modified cells were enriched in HIV-1-infected mice compared with uninfected mice (Fig. 5f). Therefore, CCR5-modified CD4+ T cells by SaCas9 showed resistance and enrichment in humanized mice.
CCR5 disruption in human CD34+ HSPCs via lentiviral vector expressing SaCas9/sgRNA
Human CD34+ HSPCs are a significant tool for gene therapy of some hereditary genetic disorders such as hematological diseases [47] because of their ability to generate a hematopoietic system. Disruption of CCR5 in human CD34+ HSPCs by ZFN can provide long-term antiviral effects [48]. Researchers also successfully used CRISPR/SaCas9 to generate a SOX2 reporter in a human induced pluripotent stem cell line [49]. Since the LentiCRISPR/SaCas9-mediated disruption of CCR5 in human primary CD4+ T cells was effective in increasing HIV-1 resistance in vitro and in vivo, we next determined whether LentiCRISPR/SaCas9 disruption of CCR5 might have the same effect in human CD34+ HSPCs. We isolated human CD34+ HSPCs from umbilical cord blood and cultured the cells with SCF, Flt-3L and TPO. LentiCRISPR-SaCas9/sgRNA-#8 or control were transfected into human CD34+ HSPCs by nucleofection, and the T7E1 assay was performed to confirm disruption of CCR5 at 72 h post-transfection. The results indicated that sgRNA-#8 induced CCR5 gene editing in human CD34+ HSPCs (Fig. 6a) and DNA sequencing demonstrated deletion and mutation of the CCR5 gene (Fig. 6b). The colony-forming unit (CFU) assay is a standard protocol to evaluate the normal differentiation potential of stem cells [33, 36]. To determine the phenotype of HSPCs development, we conducted CFU assays of non-treated and CCR5-modified CD34+ HSPCs. The numbers and types of colonies formed by control and CCR5-modified cells suggested that differentiation potential of HSPCs was not affected by CRISPR/SaCas9-mediated gene editing (Fig. 6c). Due to the small number of human CD34+ HSPCs, however, we did not conduct in vivo assays in mice with R5-modified human CD34+ HSPCs.

CCR5-editing in the human hematopoietic stem cells by LentiCRISPR/SaCas9 maintains potential multilineage ability. a CCR5 gene editing by LentiCRISPR/SaCas9-sgRNA-#8 or control in CD34+ HSPCs analyzed via T7E1 assay. b Sanger sequencing of CCR5 gene target loci in modified primary CD34+ HSPCs. c Numbers of colony types generated from untreated and CCR5-modified CD34+ HSPCs by CFU assay. Data were analyzed by unpaired t-test and error bars showed the mean ± SEM of three independent experiments
CRISPR/SaCas9-mediated CCR5 disruption is highly specific and non-toxic to cells
The type II CRISPR/Cas9 system has been widely used in recent years for various studies. Although it is a convenient and efficient approach, it may have potential off-target effects that could limit its clinical utility [50]. To test the potential off-target cleavage mediated by SaCas9/sgRNA, we compared sgRNA-#6 and #8 target sequences against the human genome to determine the potential off-target binding sites by an online search tool (http://www.rgenome.net/cas-offinder/) [51]. 10 potential off-target sites were screened (Additional file 5: Table S2) and about 800-bp PCR products from Jurkat T cell genomic DNA were tested by T7E1 assay. We did not identify indels in these potential off-target sites in Jurkat T cells edited by CRISPR/SaCa9 (Additional file 3: Fig. S3a, b), indicating the combination of sgRNA-#6 and #8 with CRISPR/SaCa9 exhibited high specificity in our experiments. When a DSB in DNA is induced, the p53 binding protein 1 (53BP1) can be recruited to the DSB site to mediate NHEJ and facilitate the repair response [30, 52]. Thus, we can quantify the number of genome-wide DSBs induced by CRISPR/SaCas9 through detection of 53BP1 foci in the nucleus. The genomic integrity of Jurkat T cells was assessed 2 days post-transduction with Lenti-SaCas9/sgRNA-#6 and #8 by observing and counting the number of 53BP1 foci per nucleus via immunostaining. Compared with etoposide-treated positive control cells, the number of 53BP1 foci per nucleus of the SaCas9/sgRNA-treated cells and untreated cells was much lower (Additional file 3: Fig. S3c). Otherwise, the MTT assay results of CRISPR/SaCas9-treated and untreated Jurkat T cells cultured in low serum medium showed no toxicity at different time points when cells were under stress (Additional file 3: Fig. S3d).
Discussion
Traditional antiretroviral therapy for the treatment of HIV-1/AIDS maintains undetectable levels of virus replication but cannot eradicate the proviral reservoir. Throughout the course of treatment, the HIV-1 provirus remains hidden but can be activated after cessation of therapy, which is followed by productive infection and severe disease progression [2, 7, 53]. It is impossible to cure HIV-1/AIDS by drug treatment alone. In addition, antiretroviral therapy has potential side-effects and is quite expensive in some under-developed countries, making it unaffordable as a long-term treatment [3, 4, 54]. Therefore, gene therapy may be a potential alternative approach to control HIV-1 replication and cure HIV-1/AIDS in the future [55, 56].
A small number of people with a 32-bp deletion of the CCR5 gene (CCR5Δ32) have shown to be resistant to HIV-1 infection [57,58,59], and transplantation of allogeneic bone marrow with homozygous CCR5Δ32 to AIDS patients has shown to be a functional cure with few side effects [29, 60]. From this evidence, the chemokine receptor CCR5 represents an optimal target for HIV-1/AIDS therapy. With limited availability of CCR5Δ32-homozygous donors and the potential for immunological rejection, direct disruption of CCR5 by gene editing is needed to advance HIV-1/AIDS gene therapy [32, 34, 61, 62]. ZFN-mediated disruption of CCR5 has shown efficacy in human CD4+ T cells and has been applied in a phase I clinical trial [13]. Earlier gene therapy approaches such as ZFN and TALEN have potential drawbacks and limitations such as low efficiency and high off-target effects. However, the type II CRISPR/Cas9 system has been developed rapidly in recent years as a powerful gene editing tool. It has the characteristics of convenience, high editing efficacy, and low off-target effects in mammalian cells [31, 32, 63,64,65]. CD4+ T cells in which CXCR4 was disrupted by CRISPR/SpCas9 or SaCas9 showed HIV-1 resistance [41, 66]. Here, we showed that CRISPR/SaCas9 combined with a sgRNA to target the CCR5 in human primary CD4+ T cells inhibited HIV-1 infection. Moreover, CCR5-modified CD4+ T cells engrafted into humanized mice exhibited a significant survival and enrichment advantage after R5-tropic HIV-1 infection compared to unmodified cells. This indicates that HIV-1 infection may place selective pressure on CCR5-modified CD4+ T cell survival. Due to the development of XGVHD in the HIV-1-infected NCG mice, whether the enriched CCR5-disrupted CD4+ T cell population could confer long-term resistance to HIV-1 infection was not assessed in our humanized mice study. In addition, the CCR5 gene could also be disrupted by CRISPR/SaCas9 in CD34+ HSPCs without deficiencies in differentiation. Therefore, CRISPR/SaCas9-mediated disruption of CCR5 may have the potential for application in HIV-1/AIDS therapy.
Although the CRISPR/Cas9 system is a powerful genome editing technology and has multiple advantages compared to other gene editing tools [67], the potential for off-target effects must be taken into consideration prior to clinical trials [17]. The tolerance of mismatches in the target sites by SpCas9/sgRNA has been observed in a previous study [68]. In terms of the CCR5 gene, off-target sites may exist at another C–C chemokin receptor, CCR2, or other genes [69]. Intriguingly, the efficient disruption of CCR5 via lentivirus expressing spCas9/sgRNA have shown negligible off-target effects in CD4+ T cells [32] and in CD34+ HSPCs [33]. Unlike the CRISPR/SpCas9 system which has a three-base PAM sequence, CRISPR/SaCas9 has a PAM sequence that consists of six bases. This longer PAM sequence may improve on-target recognition and reduce off-target rates. In our study, we also assessed potential off-target effects of the CRISPR/SaCas9 system with CCR5 sgRNA, and our data showed no detectable off-target editing at the selected potential off-target sites. The specificity of CRISPR/SaCas9 activity was further supported by direct staining for 53BP1 foci induced by DSB in the nucleus, which was used for the detection of cleavage at the most similar putative off-target sites in the genome. All the results suggest that ablation of CCR5 via the CRISPR/SaCas9 system may be a safe alternative approach for generation of HIV-1 resistance in human CD4+ T cells and CD34+ HSPCs, however, there are also potential drawbacks in targeting CCR5 alone in HIV-1/AIDS gene therapy. Disruption of the CCR5 co-receptor may pressure the R5-tropic HIV-1 strain to use another co-receptor, CXCR4, to replicate in cells [70]. In addition, disruption of CCR5 expression alone cannot cure the patients infected with R4-tropic HIV-1 as well as the patients infected with both R4- and R5-tropic HIV-1. To overcome this problem, researches on simultaneous disruption of both CXCR4 and CCR5 genes by ZFNs and CRISPR/SpCas9 in primary human CD4+ T cells had been conducted, and such strategies could protect cells from both R4- and R5-tropic HIV-1 infection [34, 71, 72]. The CRISPR/SaCas9 system developed in this work can be used in future studies to target both CXCR4 and CCR5 for broad-spectrum resistance against various HIV strains.
In our study, we chose two highly-efficient sgRNAs screened from thirteen sgRNAs in cell lines. But the efficiencies of CCR5 disruption in primary human CD4+ T cells and CD34+ HSPCs are lower than that in cell lines. This may be determined by many factors such as the specificity of sgRNA between cell lines and primary cells [33, 73] and inefficient delivery [31]. As shown in previous study, efficient delivery of CRISPR/Cas9 components into primary cells remains a major challenge for CCR5 editing [31]. The AAV vector is a safe and efficient vector in gene editing [18]. As the size of SaCas9 is smaller than commonly used SpCas9, the SaCas9/sgRNA-CCR5 system can be compatible with AAV vector [18, 41, 74]. In our study, we have difficulty in transducing sgRNA/SaCas9 delivered by AAV vectors into primary cells. Moreover, previous studies had found that inactivation of gene by AAV-SaCas9 remains a challenge in primary human cells due to issues such as special serotype and low transduction efficiency [75, 76]. Our successful delivery of SaCas9/sgRNA by lentivirus may show promise for the improvement of AAV-delivered SaCas9/sgRNA to disrupt CCR5 in HIV-1 gene therapy. Additionally, we showed that LentiCRISPR/SaCas9-mediated disruption of CCR5 was effective in CD34+ HSPCs, and whether engraftment of CCR5-edited CD34+ HSPCs into humanized mouse resistance to HIV-1 infection requires further exploration.
Conclusions
In summary, our study demonstrated that disruption of CCR5 using CRISPR/SaCas9 delivered by lentivirus in human primary CD4+ T cells resulted in T-cell prevention of HIV-1 infection and enrichment in humanized mice. The selective survival and enrichment of CCR5-modified CD4+ T cells in humanized mice challenged with HIV-1 may provide the reference for reconstitute immune function in individuals with HIV-1/AIDS. Moreover, CRISPR/SaCas9-mediated CCR5 editing in CD34+ HSPCs has no effect on cell differentiation, which may point to a safe application in transplantation of allogeneic bone marrow. Overall, by using a combination of small-sized SaCas9 and novel target sites of CCR5, this work provides an alternative approach to specifically and efficiently disrupt CCR5 in human cells and may offer a new choice for HIV-1/AIDS gene therapy in the future.
Methods
Cell lines, cell culture and transfection
HeLa cells and TZM-bl cells were maintained in DMEM (Gibco) and Jurkat T cells were maintained in RPMI 1640 (HyClone) as described in previously [77]. HeLa cells and TZM-bl cells were planted in 12-well plates and transfected with 1.0 µg AAV-CRISPR/SaCas9-sgRNA or Lenti-CRISPR/Cas9-sgRNA plasmids per well by Lipofectamine 2000 (Life Technologies) according to its instructions.
Construction of adeno-associated viral vector and lentiviral vector expressing CRISPR/SaCas9-sgRNA
The sgRNA targets were designed by online tool (http://crispr.cos.uni-heidelberg.de/). The target DNA were synthesized, annealed and inserted into PX601 plasmid (Addgene #61591) digested by BsaI (Fermentas) to generate the recombinant AAV vector expressing CRISPR/SaCas9-sgRNA. To construct lentiviral vector expressing SaCas9 and sgRNA, we firstly amplified SaCas9 from PX601 by PCR and inserted it into lentiCRISPR-v2 plasmid (Addgene #52961) to replace SpCas9. Then we inserted all these sgRNA targets into LentiCRISPR/SaCas9 digested with BsmbI (Fermentas). All adeno-associated viral and lentiviral recombinant vectors expressing SaCas9 and sgRNA were confirmed by sequencing. The oligonucleotides used for target CCR5 in the study were showed in Additional file 4: Table S1.
Isolation of human primary CD4+ T cells and CD34+ hematopoietic stem/progenitor cells
All human samples handling and experimental procedures were approved by the Experimental Ethics Committee of Wuhan University. The human whole blood samples were obtained from Wuhan Blood Center (Wuhan, China) donated by healthy people. Then we separated the peripheral blood mononuclear cells (PBMCs) by centrifugation at 200g for 15 min with Ficoll-Paque (BD) from the whole blood. For further separation and purification of human primary CD4+ T cells, we used CD4+ T cell isolation Kit (Miltenyi Biotech) according to the manufacturer’s instructions. CD4+ T cells were maintained in RPMI 1640 medium (HyClone) supplemented with 10% FBS (Gibco), 1% penicillin/streptomycin (HyClone) and human interleukin-2 (IL-2) (20 ng/ml, Peprotech) at 37 °C with 5% CO2. CD4+ T cells were stimulated in anti-CD3/anti-CD28-coated (BioLegend) culture dishes before transduction [78].
The human umbilical cord blood samples were collected from Zhongnan Hospital of Wuhan University (Wuhan, China) or Wuhan Hamilton Biotechnology (Wuhan, China). The human CD34+ HSPCs were isolated from umbilical cord blood by CD34+ MicroBead Kit (MACS, Miltenyi Biotec) according to its manufacturer’s instructions. The isolated CD34+ HSPCs were maintain in Stemspan serum-free medium II (STEMCELL Technologies) supplemented with cytokines including recombinant human stem cell factor (SCF; 100 ng/mL, PeproTech), recombinant human fms-related tyrosine kinase 3 ligand (Flt-3L; 100 ng/mL, PeproTech) and recombinant human thrombopoietin (TPO; 100 ng/mL, PeproTech).
Nucleofection of plasmids to human hematopoietic stem/progenitor cells and colony-forming unit (CFU) assay
About 5 × 106 human CD34+ HSPCs were electroporated with 3.0 µg Lenti-CRISPR-SaCas9/sgRNA or control plasmids using P3 Primary Cell 4D-Nucleofector Kit (V4XP-3024). Briefly, after centrifugation of 5 × 106 cells by 200g for 10 min, the culture medium was discarded and the cells were washed with 1xPBS for three times. Then re-suspended the cells by 100 µl Nucleofector Solution with 3.0 µg plasmids and electroporated using a Lonza Nucleofector 4D (E0-100). After nucleofection, the mixture was immediately transferred into the pre-warmed medium carefully and cultured at 37 °C with 5% CO2 for 6 h before replacement with fresh medium.
For CFU assay, about 2000 non-treated and CCR5-modified CD34+ HSPCs were seeded in methylcellulose medium (MethoCult H4435 Optimum, Stem Cell Technologies) according to the manufacturer’s instructions. 14 days after incubation at 37 °C with 5% CO2, total clone numbers were enumerated under inverted microscope.
Production of lentivirus and HIV-1 virus and cell transduction
The HEK293T cells were seeded in 10 cm plates overnight before transfected with 6.0 µg LentiCRISPR-SaCas9/sgRNA or LentiCRISPR-SaCas9 control plasmids, 3.0 µg pMD2.G and 4.5 µg psPAX2 plasmids combining with Polyethylenimine regent (PEI, Polysciences, Warrington, PA) and opti-MEM (Gibco) according to the manufacture’s instructions. The culture medium was collected after 3 days post transduction and filtered by 0.45 µm filter. The viral titer was tested by virus counter (Virocyt 2100) and stored in − 80 °C after aliquot. Jurkat T cells were transduced with lentivirus (MOI = 40) by centrifugation at 2000 rpm for 2 h with 8 µg/ml polybrene (Sigma) at 25 °C, then incubated for another 4 h in 37 °C with 5% CO2 before replacement with fresh medium. The CD4+ T cells were cultured in the anti-CD3 and CD8 coated plate with IL-2 in the medium for 36 h and then transduced with lentivirus (MOI = 100) just as Jurkat T cells. The transduced cells were cultured in fresh medium RPMI with 10% FBS for 3 days for further analysis. The R5-tropic HIV-1 virus (HIV-1YU2) was produced as previous described [71].
T7 endonuclease 1 (T7E1) assay and DNA sequencing
To measure the efficiency of CCR5 genomic mutation, we performed T7 endonuclease 1 (T7E1) assay [79] and sequencing analysis as previously described [66]. Briefly, according to the protocol of Blood & Cell Culture DNA Midi kit (Tiangen, China), genomic DNA was extracted from the modified cells and PCR amplification of CCR5 gene with a set of primers (Additional file 6: Table S3). The PCR products were purified by Gel Extraction Kit (Promage). Then we used 300 ng purified PCR products combined with 2 µl 10 × NEB buffer (New England BioLabs) and appropriate deionized H2O to make the final volume of 20 µl. The mixture was annealed to form the heteroduplexes and digested with five unites of mismatch-sensitive T7E1 (New England BioLabs) for 1 h at 37 °C [79]. The digested DNA was analyzed by 1.5% agarose gel and the editing frequency was quantified by Image J software as described previously [17]. The PCR products were also inserted into pEGM-T Easy Vector (Promega) for sequencing by a T7 primer.
Flow cytometry and western blotting
To detect the expression of CCR5 in cells edited by CRISPR/SaCas9, we collected the control-modified and CCR5-sgRNA/SaCas9 modified TZM-bl cells 3 days post transfection and washed with cold 1 × PBS for three times. Then the cells were strained with APC conjugated anti-human CCR5 antibody (Biolegend) for 30 min on ice and analyzed by flow cytometry (FACS AriaIII, BD). To count the number of CD4+ T cells in blood obtained from the mice in different time points, we firstly treated the blood samples by red blood lysis buffer (BD Biosciences) for 15 min, then stained the samples with PE conjugated anti-human CD4 antibody (BD Biosciences), APC conjugated anti-human CD3 antibody (BD Biosciences) and FITC conjugated anti-human CD8 antibody (BD Biosciences) for 30 min on ice. At last, all samples were analyzed by flow cytometry (FACS AriaIII, BD) and Flow Jo software (Treestar). For western blotting assay, we lysed the cells with lysis buffer containing 50 mM Tris-HCl pH = 7.4, 1% Triton X-100, 150 mM NaCl, 0.1% SDS, 1.5 Mm EDTA, 0.25% deoxycholate and protease inhibitor PMSF and cocktail (Roche Applied Science) on ice for 30 min. The lysates were centrifuged by 12,000 rpm for 10 min and the supernatants were mixed with 2 × SDS loading buffer incubation at 100 °C for 10 min. The proteins were detected by SDS-PAGE with anti-GAPDH (Proteintech) and anti-CCR5 antibodies (Proteintech).
Luciferase reporter assay, p24 antigen ELISA and real time PCR assay
CCR5-modified and control-modified TZM-bl cells (5 × 104) were seeded in 24-well plate one day before R5-tropic HIV-1 infection. All the cells were infected with HIV-1YU2 (MOI = 0.5) for 8 h and washed with 1 × PBS for three times before changing fresh medium. After 3 days infection, the TZM-b1 cells were collected and lysed with 100 µl lysis buffer (Promega). 20 µl of cell suspensions were used to measure luciferase activity by a BrightGlo Luciferase assay according to the manufacturer’s instruction (Promega). For Jurkat T cells and CD4+ T cells, the supernatant was collected at different days post infection and analyzed by a p24 ELISA Kit (RETRO-TEK) according to its instruction. The genomic DNA of CD4+ T cells in spleens were extracted and real time PCR assay was conducted to detected HIV-1 gag or human β-globin gene using SYBR Green PCR Master Mix (Invitrogen).
Humanized mice transplantation, assessment and HIV-1 challenge in vivo
All animal experiments were performed in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals. Animal handling and experimental procedures were approved by the Animal Experimental Ethics Committee of Wuhan University. Human primary CD4+ T cells were expanded and transduced as described above. The NOD-Prkdcem26Cd52Il2rgem26Cd22/Nju (NCG) humanized mice (10–12 weeks) were injected with human CD4+ T cells via caudal vein. Mice were divided into two groups randomly and each group had 8 mice received with 1 × 107 mock-modified or SaCas9/sgRNA-CCR5 modified human CD4+ T cells/per mice. The animals were purchased from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China) and were maintained in a defined flora animal center at college of life sciences, Wuhan university. We evaluated the number of CD4+ T cells in peripheral blood from each mouse eye socket vein post-injection by flow cytometry. 35 days after transplantations, half of the mice in each group received 1 × 105 cells/per infection with HIV-1YU2 and another half received 1 × 105 cells/per without virus infection. After HIV-1YU2 challenge, we began to collect the whole blood at different time point for three times and measured CD4+ T cell counts. All the mice were conducted by euthanization at 45 days post-infection and the spleen cells were collected by mechanical trituration method and passed through a 70 μm strainer to measure CD4+ T cell counts. SaCas9/sgRNA-induced CCR5 mutation frequency was analyzed by T7E1 assay.
Off-target analysis
The potential off-target sites were predicted by the online tool (http://www.rgenome.net/cas-offinder/) to look for the similar sequences with the 4-bp allowed mismatch (Additional file 5: Table S2). Those predicted off-target sequences were amplified by PCR of about 800-bp fragment centered near the off-target sites. The LentiCRISPR/SaCas9-modified Jurkat T cell genome were as templates and primes used in the analysis were listed in Additional file 6: Table S3. T7E1 assay was used to detect the off-target mutation.
3-(4,5-dimethylthiazol-2-yl)-2,5-dephrnyltetra-zolium bromide (MTT) assay
5 × 103 Jurkat T cells were plated in 96-well plates with 2% FBS in RPMI 1640 medium and cultured for another 0, 24, 48 and 72 h. At different time points, cells were collected for MTT assay by MTT Cell Proliferation and Cytotoxicity Aaasy Kit (Beyotime, China) according to its instruction manual. Briefly, 10 µl MTT (5 mg/ml) was added into each well and incubated at 37 °C for 4 h. Then 100 µl Formanzan solution was added into each well for dissolving. The optical density was read at 570 nm by Multiscan Spectrum (Bio-Tek).
Immunofluorescence assay
Staining for 53BP1 in the nuclear was conducted in Jurkat T cells 2 days post-transduction. The cells were fixed with 4% paraformaldehyde for 10 min at room temperature, then washed with 1 × PBS for twice and permeated with 0.5% TritonX-100 for 5 min. After blocking the nonspecific staining with 3% BSA for 30 min, cells were incubated with anti-53BP1 rabbit polyclonal antibodies (Cell Signaling Technology) diluted by 1% BSA at 4 °C overnight. The cells were incubated with Rhodamine-conjugated secondary antibody (Thermo Scientific) at room temperature for 1 h. Slides were mounted with DAPI (Thermo Scientific) to stain cell nuclei and viewed on the immunofluorescence microscope (ZEISS).
Statistical analysis
Statistical analysis was performed by Graph-Pad Prism 5.0 and statistical significant was calculated by unpaired t tests. *p < 0.05; **p < 0.01, and ***p < 0.001 represents significant differences. All experiments were repeated at least three times.
Availability of data and materials
All relevant data are within the paper and all data are fully available without restriction.
Change history
23 July 2019
Following publication of their article [1], the authors realized that they inadvertently omitted the contribution of Dr. Li Wu (Ohio State University) who commented on the manuscript at the early stage of the manuscript preparation and provided one plasmid related to this work.
References
Palella FJ Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338(13):853–60.
Katlama C, Deeks SG, Autran B, Martinez-Picado J, van Lunzen J, Rouzioux C, et al. Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet. 2013;381(9883):2109–17.
Siliciano JD, Siliciano RF. HIV-1 eradication strategies: design and assessment. Curr Opin HIV AIDS. 2013;8(4):318–25.
Durand CM, Blankson JN, Siliciano RF. Developing strategies for HIV-1 eradication. Trends Immunol. 2012;33(11):554–62.
Corbeau P, Reynes J. Immune reconstitution under antiretroviral therapy: the new challenge in HIV-1 infection. Blood. 2011;117(21):5582–90.
Sendagire H, Easterbrook PJ, Nankya I, Arts E, Thomas D, Reynolds SJ. The challenge of HIV-1 antiretroviral resistance in Africa in the era of HAART. AIDS Rev. 2009;11(2):59–70.
Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The challenge of finding a cure for HIV infection. Science. 2009;323(5919):1304–7.
Park CY, Lee DR, Sung JJ, Kim DW. Genome-editing technologies for gene correction of hemophilia. Hum Genet. 2016;135(9):977–81.
Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, Kan YW. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and beta-thalassemia. Proc Natl Acad Sci USA. 2016;113(38):10661–5.
Prakash V, Moore M, Yanez-Munoz RJ. Current progress in therapeutic gene editing for monogenic diseases. Mol Ther. 2016;24(3):465–74.
Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991;252(5007):809–17.
Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335(6069):716–9.
Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR13 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–10.
Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39(21):9283–93.
Shi B, Li J, Shi X, Jia W, Wen Y, Hu X, Zhuang F, Xi J, Zhang L. TALEN-mediated knockout of CCR15 confers protection against infection of human immunodeficiency virus. J Acquir Immune Defic Syndr. 2017;74(2):229–41.
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9(6):467–77.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23.
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–91.
Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15(5):321–34.
Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312(5996):763–7.
Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272(5270):1955–8.
Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382(6594):829–33.
Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA. 2008;105(21):7552–7.
Hunt PW, Harrigan PR, Huang W, Bates M, Williamson DW, McCune JM, et al. Prevalence of CXCR24 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J Infect Dis. 2006;194(7):926–30.
Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86(3):367–77.
Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382(6593):722–5.
Hutter G. More on shift of HIV tropism in stem-cell transplantation with CCR27 delta32/delta32 mutation. N Engl J Med. 2014;371(25):2437–8.
Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR28 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692–8.
Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, et al. HIV-1 remission following CCR29Delta32/Delta32 haematopoietic stem-cell transplantation. Nature. 2019;568(7751):244–8.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26(7):808–16.
Li C, Guan X, Du T, Jin W, Wu B, Liu Y, Wang P, Hu B, Griffin GE, Shattock RJ, Hu Q. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR31 using adenovirus-delivered CRISPR/Cas9. J Gen Virol. 2015;96(8):2381–93.
Wang W, Ye C, Liu J, Zhang D, Kimata JT, Zhou P. CCR32 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE. 2014;9(12):e115987.
Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15(5):643–52.
Didigu CA, Wilen CB, Wang J, Duong J, Secreto AJ, Danet-Desnoyers GA, Riley JL, Gregory PD, June CH, Holmes MC, Doms RW. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood. 2014;123(1):61–9.
Kang H, Minder P, Park MA, Mesquitta WT, Torbett BE, Slukvin II. CCR35 disruption in induced pluripotent stem cells using CRISPR/Cas9 provides selective resistance of immune cells to CCR35-tropic HIV-1 virus. Mol Ther Nucleic Acids. 2015;4:e268.
Xu L, Yang H, Gao Y, Chen Z, Xie L, Liu Y, et al. CRISPR/Cas9-mediated CCR36 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol Ther. 2017;25(8):1782–9.
Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2015;33(1):102–6.
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33(1):73–80.
Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34(3):328–33.
Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther. 2017;25(5):1168–86.
Wang Q, Chen S, Xiao Q, Liu Z, Liu S, Hou P, et al. Genome modification of CXCR41 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection. Retrovirology. 2017;14(1):51.
Wang Q, Liu S, Liu Z, Ke Z, Li C, Yu X, Chen S, Guo D. Genome scale screening identification of SaCas9/gRNAs for targeting HIV-1 provirus and suppression of HIV-1 infection. Virus Res. 2018;250:21–30.
Wen M, Arora R, Wang H, Liu L, Kimata JT, Zhou P. GPI-anchored single chain Fv—an effective way to capture transiently-exposed neutralization epitopes on HIV-1 envelope spike. Retrovirology. 2010;7:79.
Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74(18):8358–67.
Van Duyne R, Pedati C, Guendel I, Carpio L, Kehn-Hall K, Saifuddin M, Kashanchi F. The utilization of humanized mouse models for the study of human retroviral infections. Retrovirology. 2009;6:76.
Yuan J, Wang J, Crain K, Fearns C, Kim KA, Hua KL, Gregory PD, Holmes MC, Torbett BE. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Mol Ther. 2012;20(4):849–59.
Vanhee S, Vandekerckhove B. Pluripotent stem cell based gene therapy for hematological diseases. Crit Rev Oncol Hematol. 2016;97:238–46.
Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR48 control HIV-1 in vivo. Nat Biotechnol. 2010;28(8):839–47.
Balboa D, Weltner J, Novik Y, Eurola S, Wartiovaara K, Otonkoski T. Generation of a SOX2 reporter human induced pluripotent stem cell line using CRISPR/SaCas9. Stem Cell Res. 2017;22:16–9.
Xue HY, Ji LJ, Gao AM, Liu P, He JD, Lu XJ. CRISPR-Cas9 for medical genetic screens: applications and future perspectives. J Med Genet. 2016;53(2):91–7.
Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30(10):1473–5.
Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151(7):1381–90.
Marcellin F, Spire B, Carrieri MP, Roux P. Assessing adherence to antiretroviral therapy in randomized HIV clinical trials: a review of currently used methods. Expert Rev Anti Infect Ther. 2013;11(3):239–50.
Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med. 2011;62:141–55.
Rossi JJ, June CH, Kohn DB. Genetic therapies against HIV. Nat Biotechnol. 2007;25(12):1444–54.
Herrera-Carrillo E, Berkhout B. Potential mechanisms for cell-based gene therapy to treat HIV/AIDS. Expert Opin Ther Targets. 2015;19(2):245–63.
Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort. ALIVE Study. Science. 1996;273(5283):1856–62.
Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, et al. The role of a mutant CCR58 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2(11):1240–3.
Novembre J, Galvani AP, Slatkin M. The geographic spread of the CCR59 Delta32 HIV-resistance allele. PLoS Biol. 2005;3(11):e339.
Hutter G, Zaia JA. Allogeneic haematopoietic stem cell transplantation in patients with human immunodeficiency virus: the experiences of more than 25 years. Clin Exp Immunol. 2011;163(3):284–95.
Badia R, Riveira-Munoz E, Clotet B, Este JA, Ballana E. Gene editing using a zinc-finger nuclease mimicking the CCR61Delta32 mutation induces resistance to CCR61-using HIV-1. J Antimicrob Chemother. 2014;69(7):1755–9.
Mussolino C, Alzubi J, Fine EJ, Morbitzer R, Cradick TJ, Lahaye T, Bao G, Cathomen T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014;42(10):6762–73.
Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10(10):957–63.
Liu Y, Zhao M, Gong M, Xu Y, Xie C, Deng H, Li X, Wu H, Wang Z. Inhibition of hepatitis B virus replication via HBV DNA cleavage by Cas9 from Staphylococcus aureus. Antiviral Res. 2018;152:58–67.
Xiao QQ, Guo DY, Chen SL. Application of CRISPR/Cas9-based gene editing in HIV-1/AIDS therapy. Front Cell Infect Microbiol. 2019;9:69.
Hou P, Chen S, Wang S, Yu X, Chen Y, Jiang M, Zhuang K, Ho W, Hou W, Huang J, Guo D. Genome editing of CXCR66 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci Rep. 2015;5:15577.
Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, Wang Y, Brodsky RA, Zhang K, Cheng L, Ye Z. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15(1):12–3.
Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N, Wile BM, Vertino PM, Stewart FJ, Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42(11):7473–85.
Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR69 genes have substantial off-target activity. Nucleic Acids Res. 2013;41(20):9584–92.
Fatkenheuer G, Nelson M, Lazzarin A, Konourina I, Hoepelman AI, Lampiris H, et al. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N Engl J Med. 2008;359(14):1442–55.
Liu Z, Chen S, Jin X, Wang Q, Yang K, Li C, et al. Genome editing of the HIV co-receptors CCR71 and CXCR71 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci. 2017;7:47.
Yu S, Yao Y, Xiao H, Li J, Liu Q, Yang Y, Adah D, Lu J, Zhao S, Qin L, Chen X. Simultaneous knockout of CXCR72 and CCR72 genes in CD4+ T cells via CRISPR/Cas9 confers resistance to both X4- and R5-tropic human immunodeficiency virus type 1 infection. Hum Gene Ther. 2018;29(1):51–67.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–32.
Sather BD, Romano Ibarra GS, Sommer K, Curinga G, Hale M, Khan IF, et al. Efficient modification of CCR1 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. 2015;7(307):307ra156.
Chen S, Yu X, Guo D. CRISPR-cas targeting of host genes as an antiviral strategy. Viruses. 2018;10(1):40.
Deng Q, Chen Z, Shi L, Lin H. Developmental progress of CRISPR/Cas9 and its therapeutic applications for HIV-1 infection. Rev Med Virol. 2018;28(5):e1998.
Liu Y, Zhou J, Pan JA, Mabiala P, Guo D. A novel approach to block HIV-1 coreceptor CXCR77 in non-toxic manner. Mol Biotechnol. 2014;56(10):890–902.
Wilen CB, Wang J, Tilton JC, Miller JC, Kim KA, Rebar EJ, et al. Engineering HIV-resistant human CD4+ T cells with CXCR78-specific zinc-finger nucleases. PLoS Pathog. 2011;7(4):e1002020.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–84.
Acknowledgements
We thank Dr. Feng Zhang (Broad Institute of MIT and Harvard, USA) for sharing the SaCas9 plasmid. We also thank Mrs. Victoria V. Maksimova (Ohio State University) and Dr. Karthik M. Kodigepalli (Ohio State University) for helping with revising the manuscript.
Funding
This work was funded by the China National Special Research Program of Major Infectious Diseases (#2017ZX10202102) and Hubei Provincial Science & Technology Innovation Team Grant (#2012FFA043). D.G. is supported by Guangdong Provincial “Zhujiang Talents Program” and the National Ten-Thousand Talents Program. This work was also supported by the Natural Science Foundation of China (81401659), China Postdoctoral Science Foundation (2015T80838 and 2014M560622) and the scholarship from the China Scholarship Council. to S.C.
Author information
Authors and Affiliations
Contributions
DG conceived and designed the experiments. QX, SC, QW, ZL, SL, HD performed the experiments. WH, YX provided key suggestions. DW, JL provided HSC. QX, SC, DG analyzed the data. QX, SC, DG wrote and revised the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were performed in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals. Animal handling and experimental procedures were approved by the Animal Experimental Ethics Committee of Wuhan University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Fig. S1.
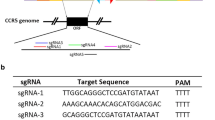
The schematic of CCR5 gene targeted locus in this study. The CCR5 gene locates at the short arm of chromosome 3 and the open reading frame (ORF) of CCR5 is in the fourth exon with the base pair from 46,370,854 to 46,376,206 referred to GRCh38 coordinate. 13 sgRNAs were designed by protospacer adjacent motif (PAM) with 5′-NNGRRT-3′ or 3′-NNCYYA-5′ sequences.
Additional file 2: Fig. S2.
Screening of the effective CCR5-sgRNAs in HeLa cells by the T7E1 assay. HeLa cells were seeded in 12-well plates and transfected with 1 µg AAV-Cas9/sgRNAs. T7E1 assay was conducted in AAV-SaCas9/sgRNA modified cells 3 days post transfection. Neg: CXCR4-sgRNA; Con: AAV vector only; #1–#13: AAV vector expressing SaCas9/sgRNA-#1–#13.
Additional file 3: Fig. S3.
Off-target analysis of CCR5-sgRNA-#6 (a) and #8 (b) in Jurkat T cells by T7E1 assay. (c) Detection of 53BP1 localization in the cell nucleus by immunostaining and epifluorescence microscopy 2 days after Jurkat T cells were transduced with lentivirus expressing SaCas9/sgRNA. Untreated cells as a negative control and 1 µM etoposide treated cells as a positive control. The consensus scale bar was 20 µm. (d) MTT assay to measure the cell viability at 0, 24, 48 and 72 h in low serum medium after transduced with lentivirus expressing SaCas9/sgRNA for 3 days in Jurkat T cells. Data were analyzed by unpaired t-test and error bars showed the mean ± SEM of three independent experiments.
Additional file 4: Table S1.
Oligonucleotides of sgRNA for targeting CCR5 locus.
Additional file 5: Table S2.
List of potential off-target sites.
Additional file 6: Table S3.
Primers used in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xiao, Q., Chen, S., Wang, Q. et al. CCR5 editing by Staphylococcus aureus Cas9 in human primary CD4+ T cells and hematopoietic stem/progenitor cells promotes HIV-1 resistance and CD4+ T cell enrichment in humanized mice. Retrovirology 16, 15 (2019). https://doi.org/10.1186/s12977-019-0477-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-019-0477-y