
Abstract
Background
Atypical antipsychotic agents, such as clozapine, are used to treat schizophrenia and other psychiatric disorders by a mechanism that is believed to involve modulating the immune system. Multiple sclerosis is an immune-mediated neurological disease, and recently, clozapine was shown to reduce disease severity in an animal model of MS, experimental autoimmune encephalomyelitis (EAE). However, the mode of action by which clozapine reduces disease in this model is poorly understood.
Methods
Because the mode of action by which clozapine reduces neuroinflammation is poorly understood, we used the EAE model to elucidate the in vivo and in vitro effects of clozapine.
Results
In this study, we report that clozapine treatment reduced the infiltration of peripheral immune cells into the central nervous system (CNS) and that this correlated with reduced expression of the chemokines CCL2 and CCL5 transcripts in the brain and spinal cord. We assessed to what extent immune cell populations were affected by clozapine treatment and we found that clozapine targets the expression of chemokines by macrophages and primary microglia. Furthermore, in addition to decreasing CNS infiltration by reducing chemokine expression, we found that clozapine directly inhibits chemokine-induced migration of immune cells. This direct target on the immune cells was not mediated by a change in receptor expression on the immune cell surface but by decreasing downstream signaling via these receptors leading to a reduced migration.
Conclusions
Taken together, our study indicates that clozapine protects against EAE by two different mechanisms; first, by reducing the chemoattractant proteins in the CNS; and second, by direct targeting the migration potential of peripheral immune cells.
Similar content being viewed by others


Background
Multiple sclerosis (MS) is an autoimmune disease characterized by continuous infiltration of autoreactive T cells and other inflammatory immune cells from the periphery into the central nervous systems (CNS). The clinical symptoms of MS are attributed to the inflammatory lesions in the CNS white matter regions leading to sensory dysfunction and loss of motor control. During MS, the pathological recruitment of myelin-specific CD4 T cells and other immune cells into the CNS results in demyelination of the neuronal axons [1, 2]. This immune cell infiltration and demyelination can be studied in the well-established experimental autoimmune encephalomyelitis (EAE) MS animal model [3]. Current therapeutic strategies including glatiramer acetate, interferon-β, or natalizumab show only limited effects on the different forms of MS [4,5,6,7]. Most of these medicines are immunomodulatory agents acting in the circulating compartment due to the low capacity to cross the intact blood-brain barrier [8, 9].
Under physiological conditions, the blood-brain barrier protects the CNS against leukocyte infiltration. Understanding which factors regulate the initial and early infiltration into the CNS during MS is central to the development of treatment strategies; however, the specific factors that regulate leukocyte trafficking and accumulation in the CNS are not fully defined [10]. Migration of activated leukocytes and macrophages is controlled by several different proinflammatory chemotactic cytokines, called chemokines [11]. These are produced and released locally and diffuse into the bloodstream thereby attracting leukocytes to the site of inflammation. Monocyte chemoattractant protein 1 (MCP-1) or CCL2 and regulated upon activation normal T cell expressed and secreted (RANTES) or CCL5 belong to the family of C-C chemokines involved in the recruitment of monocytes, macrophages, and activated lymphocytes to the site of expression [12] and mediate leukocyte adhesion to epithelial cells [13]. CCL2 and CCL5 are expressed by a wide array of different cell types constitutively within the CNS or secreted by infiltrating blood-derived macrophages upon their migration into the CNS. Interestingly, the production of CCL2, CCL3, and CCL5 in the CNS has been associated with acute disease symptoms in rat and mice [14, 15]. Given the evidence that CCL2 and CCL5 are highly involved in the regulation of EAE, modulating the expression or function of CCL2 and CCL5 attracts much attention as a potential therapy for MS [16, 17]. Drugs that would target CCL2 and CCL5 expression directly would be highly beneficial.
Clozapine is a small molecule drug that readily crosses the blood-brain barrier [18], and is an atypical antipsychotic agent used in the treatment of neuropsychiatric disorders like schizophrenia [19]. Neuropsychiatric disorders are increasingly recognized as being associated with inflammation with elevated expression of inflammation markers in the CNS. The atypical antipsychotic drug clozapine has been shown to be effective at reducing disease in EAE in a prophylactic [20] and therapeutic manner [21]. The ability of clozapine to reduce disease severity, however, was found not to be mediated by a direct effect on myelin-specific CD4 T cells [22]. Therefore, a different mechanism of action is believed to underlie the beneficial effects of clozapine treatment in EAE.
In this study, we investigated the underlying mechanism by which clozapine reduces disease onset and severity by evaluating the initial infiltration of immune cells into the CNS to determine if immune cell migration is the functional target of clozapine.
Methods
Animals
Female C57BL/6 J mice were bred and housed in the animal facility at Victoria University of Wellington, New Zealand and used between 8 and 12 weeks of age.
Ethics statement
All of the experiments with animals were carried out in the School of Biological Sciences Animal Facility at Victoria University of Wellington and were approved by the Victoria University of Wellington Animal Ethics Committee (2014-R23).
EAE induction and treatments
Mice were immunized s.c. in the rear flanks with myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (50 μg/mouse; Genescript, Piscataway, NJ) in complete Freund’s adjuvant (Sigma, St. Louis, MO) containing 500 μg/mouse Mycobacterium tuberculosis (Fort Richard, Auckland, New Zealand). In addition, mice were injected i.p. with pertussis toxin (200 ng/mouse; List Biochemicals, Campbell, CA) on days 0 and 2. Mice were weighed and scored daily as follows: 0, normal; 1, partial tail paralysis; 2, full tail paralysis; 3, paralysis in one hind limb; 4, paralysis in both hind limbs; and 5, moribund. One day before immunization, mice drinking water was changed to 60 mg/kg/day Clozapine (kindly supplied by Douglas Pharmaceuticals Ltd. (Auckland, New Zealand)) or vehicle (0.1 M acetic acid) in the drinking water. Mice were treated with clozapine or vehicle over the whole time of the experiment. Following CO2 euthanasia, brains, spinal cord, spleens, and blood were isolated and processed into a single-cell suspension.
Primary cell isolation into single-cell suspension
Spinal cord was minced and incubated with collagenase type II for 30 min at 37 °C. Remaining clumps were broken up using at pipette and cell suspension was passed through a 70 μm cell strainer and centrifuged at 760 g for 5 min. Brain was mashed through a 70 μm cell strainer and centrifuged at 760 g for 5 min. Spinal cord and brain cell pellet were resuspended in 37% Percoll™ gradient and centrifuged 30 min at 760 g without brakes. Myelin layer was removed, supernatant discharged, and pellet resuspended for cell counting. Lymph nodes were mashed through a 70-μm cell strainer and centrifuged at 760 g for 5 min and cell pellets were resuspended for cell counting.
Spleen was mashed through a 70-μm cell strainer and centrifuged at 760 g for 5 min, pellet was loosen and resuspended in Red Cell Lysis buffer for 2 min. Whole blood was also incubated with Red Cell Lysis buffer for 2 min, wash buffer was added, and samples were centrifuged at 760 g for 5 min. For the blood, lysis step was repeated at least once. Afterwards, cell pellets were resuspended for cell counting.
Flow cytometry
For the detection of immune cells, the following antibodies were used: CD4-BV421 (RM4-5; Biolegend, San Diego, CA, USA), CD45-BV510 (30-F11; Biolegend), CD25-AF488 (PC61; Biolegend), CD8-PerCPCy5.5 (53-6.7; Biolegend), CD11b-PE-Cy7 (M1/70; Biolegend), CD3-APC-Cy7 (17A2; Biolegend), Ly6C-PE (HK1.4; Biolegend), Ly6G-APC (1A8; Biolegend), CD45R-AF488 (RA3-6B2; BD Bioscience), CCR2-PE (475,301; R&D), CCR5-APC (HM-CCR5; Biolegend), and Gr1-APC-Cy7 (RB6-8C5, Biolegend). Cells were incubated with Fc Block (2.4G2; BD Biosciences) for 15 min prior to staining with fluorescently labeled antibodies for 30 min on ice. Flow cytometry was performed on a BD FACS Canto II (BD Biosciences) and analyzed using Flowjo software version 10.1 (Treestar Inc., Ashland, OR, USA).
In-vivo migration assay
C57BL/6 mice were treated with 60 mg/kg/day Clozapine (kindly supplied by Douglas Pharmaceuticals Ltd. (Auckland, New Zealand)) or vehicle (0.1 M acetic acid) in the drinking water for 7 days. On the following day, mice were injected s.c. with either 10 μg/ml of the chemokine CCL5 (RANTES; Peprotech) or 1 μg/ml of the chemokine CCL2 (MCP-1, Peprotech) in 50 μl dPBS (Invitrogen, USA) into the left hind flanks of the mice, while an equal volume of dPBS (vehicle) was injected into the right hind flank. Eighteen hours following hind flank injections, cells from the draining lymph nodes were isolated, counted, and processed for flow cytometry analysis, as described above.
Isolation and in-vitro culture of cells
Primary microglia (pooled from four to five mice per experiment) were derived as described previously [23], purity was checked by flow cytometry staining for CD45lowCD11b+CD3− and was on average 76%. Primary microglia were seeded at 5 × 104 cells/well in microglia media and 10 ng/ml macrophage colony-stimulating factor (M-CSF) (ProSpec) in a flat-bottomed 96-well plate and cultured for 4 weeks with media changes every 3–4 days. After 4 weeks in culture, the mature, adherent microglia in the 96-well plates were stimulated with or without LPS (200 ng/ml, Sigma) and pre-treated or co-treated with 20 μM clozapine.
Bone marrow-derived macrophages (BMMO) were isolated and cultured as described [24]. BMMO were either derived with 5 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech) and 5 ng/ml IL3 (Peprotech) or 10 ng/ml M-CSF for 9 days (ProSpec). BMMO (105/well) were cultured in complete culture media in 96-well plates. GM-CSF-derived BMMO were primed overnight with interferon gamma (IFNγ) (20 U/ml; Peprotech) before stimulating with or without LPS (200 ng/ml, Sigma) and 20 μM or 40 μM clozapine. M-CSF-derived BMMO were primed overnight with IL4 (20 ng/ml; ProSpec) before stimulating with or without LPS (200 ng/ml, Sigma) and 20 μM or 40 μM clozapine.
Primary astrocytes were kindly generated and provided by Matt Rowe (Victoria University of Wellington) using the Neural Tissue Dissociation Kit (T) (Miltenyi Biotech) as per supplied instructions. Microglia, oligodendrocytes, and neurons were sitting on top of the bottom layer of astrocytes. Cells were cultured in specific astrocyte medium. Microglia-containing supernatant and oligodendrocytes/neurons-containing supernatant was mechanically shaken off and discarded. Adhered cells were approximately 90% astrocytes. Cells were stimulated with IFNγ (20 U/ml; Peprotech) overnight before stimulating with or without LPS (200 ng/ml, Sigma) and 20 μM or 40 μM clozapine.
After the treatment, supernatant was frozen at − 20 °C and cells were collected and re-suspended in TRIZOL for RNA analysis and frozen at − 80 °C.
In-vitro migration assay
To assess cell motility, 120,000 GM-CSF/IL3-derived BMMO were seeded in complete culture medium into wells of the 96-well ORIS plate (Platypus Technologies, WI, USA) containing a round silicon insert set in each well. Cells were incubated overnight to settle before plugs were removed with the supplied tool. Media was carefully aspirated and replaced with fresh media. Each well was inspected using the microscope, noting wells with disturbed exclusion zones: these wells were not used. Images of wells were captured at this time to measure original wound size and used as timepoint 0. Cells were either left unstimulated, stimulated with 25 μM clozapine, or 0.001 μM Latrunculin A (Sigma-Aldrich) as a positive control or the respective vehicles. Plates were incubated for 3 days before images of each well were taken to measure final wound size. Cell viability was assessed via MTT assay at the end of the experiment.
ImageJ running the MRI Wound Healing Tool macro (Montpellier RIO Imaging, Montpellier, France) was used to measure wound size from images using the following script parameters: method: variance; variance filter radius: 5; threshold: 50; radius open: 1; min. size: 10,000. Compound-induced changes to wound closure were assessed using the following equations:
RNA analysis
Total mRNA was isolated by re-suspending the cell pellets in TRIZOL reagent and mRNA extraction was performed by using the Direct-zol™ RNA MiniPrep (Zymo Research) according to manufacturer’s protocol. In summary, TRIZOL cell suspension was mixed in 1:1 ratio with 100% absolute ethanol and added on Zymo-Spin™ IIC columns where mRNA bounds to membrane. Column was washed and DNA was denatured by DNase I digestion on the column. After washing with two buffers with decreasing salt concentration, mRNA was eluted in 30 μl RNase and DNase free water and stored at − 80 °C for further analysis.
Concentration of total mRNA was determined by measuring absorbance at 260 nm using the Nanodrop 2000 (Thermo Fischer). For cDNA generation, 100 ng of total mRNA was used and transcribed with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed using the Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen) in combination with the CFX Connect™ Real-Time PCR Detection System (BioRad). Relative quantification was performed by normalizing to the reference gene Cyclophilin A and to healthy vehicle control by using the 2–ΔΔCT (Livak) Method.
Analysis of cytokines by Milliplex
Mouse brains were isolated, weight, and mashed in RIPA Buffer (500 mg/ml) using a tissue homogenizer. Samples were incubated shaking 30 min at 4 °C, centrifuged 10 min at 10,000 g at 4 °C, and supernatant was analyzed for cytokine expression using the 32-plex MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel (MCYTOMAG-70K Millipore, Germany) according to manufacturer’s protocol. Briefly, single magnetic beads coupled with specific cytokines were mixed. Then, 50 μl of mixture was added to a 96-well plate and washed twice with Washing Buffer by using the BioPlex Pro™ II Wash Station (BioRad) with the magnetic plate carrier. Further, 50 μl of cytokine standards and samples were loaded on the plate and were incubated for 30 min at RT vertical shaking at 300 rpm by using IKA MTS 2/4 digital shaker (IKA WERKE). Samples and standards were removed by the Wash Station and wells were washed three times with washing buffer. Single specific detection antibodies for each cytokine were mixed and 25 μl of mixture was added to all wells followed by incubation for 30 min at RT vertical shaking at 300 rpm. Before adding 50 μl secondary streptavidin-PE conjugated antibody, wells were washed three times. Incubation with streptavidin-PE was performed for 10 min at RT vertical shaking at 300 rpm followed by three times washing with washing buffer. Further, 125 μl of Assay Buffer was added to each well and plate was shaked vertical for 30 s at 600 rpm. Data acquisition was performed by using the BioPlex 200 System and data analysis by using the BioPlex Manager™ software.
Monocyte isolation and enrichment
Single cell suspensions from spleens were generated as described above. Monocyte isolation was performed using the EasySep™ Mouse Monocyte Isolation Kit (StemCell Technology) according to manufacturer’s instructions. Briefly, samples were incubated with rat serum before the selection cocktail and the RapidSpheres™ were added. After the incubation time, samples were placed into the magnet and the enriched cell suspension was poured out in a continuous motion. The enriched cell suspension was again placed into the magnet and the higher enriched cell suspension was collected in the same way. Isolated cell purity was check by flow cytometry staining for CD45, CD3, and CD11b. Cells were counted as monocytes that were CD45 and CD11b positive and CD3 negative, purity was 70–90%. Cells were plated at 1 × 105 cells/well in a 96-well plate and used for cAMP measurement.
Measuring intracellular cyclic AMP
Primary splenocytes were isolated into a single cell suspension and seeded onto a 96-well plate at 1 × 106 cells/well. Splenocytes and isolated monocytes were incubated with stimulation buffer containing IBMX for 30 min prior of stimulation. Cells were either pre-treated with 20 μM clozapine for 1 h following forskolin (6.25 μM, Sigma), CCL2, or CCL5 (100 ng/ml; Peprotech) treatment or co-stimulated with the other treatments for 18 h. Monocyte in addition were co-stimulated with LPS (200 ng/ml, Sigma) for the whole time. Cells were washed with phosphate-buffered saline (PBS) and dried in pure ethanol. Afterwards, lysis buffer was added for 1 h shaking. Cell lysates were stored at − 20 °C until further use. Intracellular cyclic AMP levels were measured using the AlphaScreen Kit (Perkin Elmer) according to manufacturer’s instructions. Briefly, in an OptiPlate (Perkin Elmer), 5 μl acceptor beads were added per well and incubated with 5 μl standards and samples for 30 min prior to adding 15 μl donor beads for 8–12 h in reduced light conditions at room temperature. Plate was measured on an EnSpire multilable plate reader (Perkin Elmer) using standard AlphaScreen settings.
Statistical analyses
All graphs and statistical analyses were generated using GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA USA). Comparisons between two groups were performed using a paired Student’s t test. For comparison of more than two groups, one-way or two-way analysis of variance (ANOVA) was used with the recommended multiple comparison tests as indicated in the figure legend and as recommended by GraphPad Prism. Differences of p < 0.05 were considered significant.
Results
Clozapine reduces infiltration into the CNS
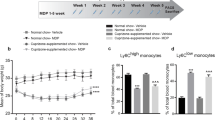
Clozapine has been shown to reduce disease severity in the EAE model of MS when administered prophylactically or therapeutically [20, 21]; however, the mechanism of protection is not clear. To understand if clozapine altered the early infiltration of immune cells into the CNS post immunization, female C57BL/6 mice were treated with clozapine or vehicle commencing 1 day prior to immunization throughout the whole course of the experiment and the number and type of immune cells in the spinal cord and brain were analyzed by flow cytometry 5, 7, 9, and 11 days after EAE induction (Additional file 5: Figure S5a,b).). As expected, the onset of disease occurred 10 days after EAE induction in the vehicle-treated animals while clozapine-treated animals showed no overt signs of disease following EAE induction (Fig. 1a, b). Microglia numbers in the spinal cord and brain did not change over time or with different treatments (Fig. 1c, g). In the spinal cord, infiltrating neutrophils were observed at day 5 in EAE mice whereas clozapine treatment significantly reduced neutrophil cell numbers (Fig. 1e). Additionally, monocyte and neutrophil cell numbers were reduced in the spinal cords and brains of clozapine-treated EAE animals compared to vehicle over the whole time of observation (Fig. 1d, e, h, i). T cell infiltration was significantly reduced in clozapine-treated EAE mice compared to vehicle at day 11 in the spinal cord as well as in the brain (Fig. 1f, j). Together, these results indicate that clozapine reduces the infiltration of monocytes, neutrophils, and T cells pre and post disease onset.

Clozapine treatment reduces infiltration into the CNS during the onset of EAE. C57BL/6 female mice were treated with clozapine (60 mg/kg/day) or vehicle control in their drinking water commencing one day prior to immunization and were scored daily (a, b). At day 5, 7, 9, and 11 after EAE induction spinal cord (c–f), brain (g–j), spleen (k–n), and blood (o–r) was collected and cell numbers were analyzed by flow cytometry. Shown are the means and SEM of individual mice (n = 9/treatment group) from three independent experiments normalized to healthy vehicle for each day. *p < 0.0332, **p < 0.021, and ***p < 0.0001 by two-way ANOVA with Tukey’s multiple comparisons test comparing EAE vehicle versus EAE Clozapine. ###p < 0.0002 and ###p < 0.0001 by three-way ANOVA comparing healthy versus EAE
To determine if the reduction in immune cell infiltration into the CNS correlated to immune cells in peripheral sites, similar assessments were done in whole blood and isolated splenocytes. Both monocytes and neutrophils numbers in the spleen and blood were elevated by EAE and were significantly reduced in the spleen in clozapine-treated EAE mice compared to vehicle (Fig. 1l, m). In contrast to the CNS, macrophage and T cell numbers were not altered by time, treatment, or immunization (Fig. 1k, n, o, r). Aside from a reduction in monocyte and neutrophil recruitment to the spleen by clozapine treatment, cell numbers of macrophages and T cells were not significantly altered in the spleen or blood pre and post EAE onset.
Clozapine reduces the expression of CCL2 and CCL5 in the CNS
Because these results indicate that upon clozapine treatment, less immune cells infiltrate into the CNS during the early period of disease onset, we investigated the expression of chemokines and cytokines in the CNS. Applying a 32-plex for the analysis of 32 different cytokines known to be involved in EAE [10, 25, 26], we found that only a few of the analyzed cytokines were regulated by EAE and clozapine treatment (Additional file 1: Figure S1e). Two chemokines, CCL2 and CCL5, which showed already an upregulation at the early timepoints were analyzed in more detail. These two cytokines are also known to recruit monocytes, T cells, and neutrophils to the sites of inflammation, and are known to be important in CNS inflammation during EAE [12, 13]. Analysis of CCL2 and CCL5 mRNA expression over time revealed that both transcripts increased in the spinal cords of vehicle-treated EAE compared to healthy animals by 11 days post-immunization and CCL2 and CCL5 levels were reduced by clozapine treatment (Fig. 2a,b). In the brain, CCL2 mRNA was increased at 7 days post-immunization and then returned to normal, while CCL5 mRNA was increased at 11 days post-immunization. As in the spinal cord, treatment with clozapine reduced the levels of chemokine expression to that of healthy control animals (Fig. 2c, d). The reduced chemokine expression of mRNA in the brain was also reflected in the expression of the protein where a significant difference between vehicle and clozapine-treated EAE animals was detected day 11 post EAE induction (Fig. 2e, f). Although EAE did significantly increase CCL2 expression in the blood 11 days post immunization, clozapine treatment did not alter it (Additional file 2: Figure S2c). No difference in CCL5 mRNA expression was detected in the blood (Additional file 2: Figure S2d) and for both chemokines in the spleen (Additional file 2: Figure S2a, b) of healthy and EAE mice treated either with vehicle or clozapine. Overall, the reduction in EAE-induced CCL2 and CCL5 in the spinal cord and brain but not spleen or blood parallels the reduction in immune cell recruitment into the CNS but not peripheral sites in clozapine-treated animals.

Clozapine treatment decreases mRNA and protein CCL2 and CCL5 expression in the CNS during the onset of EAE. C57BL/6 female mice were treated with clozapine or vehicle control in their drinking water. At day 5, 7, 9, and 11 after EAE induction spinal cord and brain was collected. a–d RNA was extracted and analyzed by qRT-PCR for CCL2 and CCL5 expression. Shown are the means and SEM of individual mice (n = 9/treatment group) from three independent experiments normalized to cyclophilin A as a housekeeper and healthy vehicle for each day. e, f Brains were lysed and protein expression was analyzed by Milliplex. Shown are the means and SEM of individual mice (n = 3/treatment group) normalized to healthy vehicle for each day. *p < 0.0332, **p < 0.021, ***p < 0.0002, and ****p < 0.0001 by two-way ANOVA with Tukey’s multiple comparisons test
Resident microglia and macrophages are targeted by clozapine for reduced chemokine expression
To test if clozapine reduces the expression of CCL2 and CCL5 in the CNS by directly targeting the resident microglia and to confirm which cells may be the source of these chemokines, primary microglia were derived as described previously [23] and astrocyte-rich cultures were isolated using the Neural Tissue Dissociation Kit (T), and activated by LPS in the presence or absence of clozapine. Upon activation of microglia, a high expression of CCL2 and CCL5 mRNA was detected (Fig. 3a, b). The expression of CCL2 was reduced if the microglia were pre- or co-treated with clozapine (Fig. 3a) but CCL5 was only significantly reduced by co-treatment (Fig. 3b). To investigate the effect of the activation status of macrophages and their role in cytokine secretion in EAE and in response to clozapine treatment, classically (M1-like) or alternatively (M2-like) activated bone marrow-derived macrophages were generated. Similar results were observed in these macrophages. Activation of these macrophages by either IFNγ and LPS or IL4 and LPS increased the expression of CCL2 and CCL5 mRNA while clozapine treatment reduced the expression (Fig. 3c–f). In contrast, while primary astrocytes showed an increased expression of CCL2 and CCL5 mRNA upon activation with LPS and IFNγ, treatment with clozapine had no effect on CCL2 or CCL5 expression at the time of observation (Fig. 3g, h). These findings indicate that microglia and macrophages present in the CNS at the time of disease induction may be directly targeted by clozapine leading to reduced activation of these cells [21], reduced chemokine expression, and consequently, changing infiltration into the CNS.

Clozapine treatment reduces CCL2 and CCL5 mRNA expression by primary macrophages and microglia. a, b Primary microglia were isolated from the brains of 5 days old mice, differentiated with M-CSF for 30 days and pre or co-treated with clozapine in the presence of LPS for 24 h. c–f Bone marrow-derived macrophages were isolated, differentiated with either GM-CSF and IL3 (c, d) or M-CSF (e, f) for 9 days and treated with clozapine in the presence of either IFNγ and LPS (c, d) or IL4 and LPS (e, f) for 24 h. g, f Primary astrocytes were isolated from brains and treated with clozapine in the presence of IFNγ and LPS for 24 h. For all samples, mRNA was extracted and analyzed by qRT-PCR for CCL2 and CCL5 expression. Shown are the means and SEM of three independent experiments normalized to cyclophilin A as a housekeeper and untreated control. *p < 0.0332 and ***p < 0.0001 by paired one-way ANOVA with Sidak’s multiple comparisons test
Clozapine inhibits migration in the presence of excessive chemokines
To verify if the reduced migration into the CNS is due solely to reduced chemokine expression, or if migration is directly altered by clozapine, in-vitro migration experiments were performed. The Oris wound healing system was used wherein GM-CSF/IL3-derived bone marrow-derived macrophages were seeded in wells containing a plug. After settlement of the cells, the plug was removed to leave a “wound” or specific area free of cells. Macrophages were unstimulated or stimulated with clozapine, Latrunculin A, or respective vehicle. As shown in Fig. 4a and Additional file 3: Figure S3a, bone marrow-derived macrophages left untreated or treated with the respective vehicles migrate to close the wound. As expected, migration was significantly inhibited by Latrunculin A, which is known to inhibit migration by preventing actin polymerization. Interestingly, clozapine also significantly inhibited the migration of macrophages in this assay (Fig. 4a and Additional file 3: Figure S3a) showing that migration can be directly inhibited by clozapine without any other stimulatory factors.

Clozapine treatment reduces CCL2- and CCL5-mediated migration. a In-vitro migration assay of bone marrow-derived macrophages treated with Latrunculin A or clozapine. b–i C57BL/6 female mice were treated with clozapine or vehicle control in their drinking water for 7 days. At the last day, CCL2 or CCL5 (left hind flank) or PBS (right hind flank) was injected s.c., and 18 h later, the draining LN cells were isolated and analyzed by flow cytometry. The total number of LN cells after CCL2 (b) or CCL5 injection (f) and of the individual cell types in the LN after CCL2 (c–e) or CCL5 injection (g–i) from three independent experiments (n = 13–14/group) are shown. P—PBS, 2—CCL2, 5—CCL5, *p < 0.0332 and **p < 0.021 by paired one-way ANOVA with Sidak’s multiple comparisons test within vehicle and clozapine treated groups. Comparison between CCL treatment in vehicle and clozapine mice was done by two-way ANOVA with Tukey’s multiple comparison test #p < 0.0332, ##p < 0.021, ###p < 0.0002, and ####p < 0.0001
To analyze if in-vivo chemokine-mediated migration is also directly inhibited by clozapine in the presence of excessive chemokine, an in-vivo migration assay was performed. To this end, CCL2 (abbreviated in Fig. 4 as “2”) or CCL5 (abbreviated in Fig. 4 as “5”) was injected s.c. in the lower left hind limb of mice, and PBS (abbreviated in Fig. 4 as “P”) was injected s.c. in the right lower hind limb as a control. Eighteen hours later, the draining lymph nodes were collected and analyzed by flow cytometry to enumerate and phenotype the immune cells (Additional file 6: Figure S6a). To determine the effect of clozapine, animals were treated with vehicle or clozapine for 7 days prior to administration of CCL2 or CCL5. Injection of CCL2 in vehicle-treated animals significantly increased cell numbers in the draining lymph nodes (Fig. 4b) compared to PBS injection, as expected. Interestingly, clozapine treatment significantly abolished this effect with similar cell numbers detected in the lymph nodes draining the site of CCL2 or PBS injection (Fig. 4b). Analysis of the cellular populations showed that monocytes, neutrophils, and T cells were significantly increased in the CCL2-affected lymph nodes of vehicle-treated mice, whereas the cellular populations in the lymph nodes were similar between PBS or CCL2 injection in clozapine-treated animals (Fig. 4c–e, Additional file 3: Figure S3b-f).
In contrast to CCL2, CCL5 injection did not induce a significant increase in total cells in the draining lymph nodes in the vehicle-treated animals and similar numbers were found in clozapine-treated animals (Fig. 4f). When the individual cellular populations were assessed, a significant increase in the number of neutrophils was detected in vehicle-treated mice injected with CCL5 (Fig. 1h) while other cell populations were constant (Fig. 4g, i and Additional file 3: Figure S3g-k). Additionally, the increase in neutrophils was not observed in clozapine-treated mice (Fig. 4h). This result reveals that neutrophils are the main cell type responding to CCL5 in this model (Fig. 4 h). Overall, these findings indicate that clozapine reduces chemokine-induced migration of immune cells in-vivo by directly targeting the migratory potential of the immune cells.
Chemokine receptors are not altered by clozapine
To evaluate how clozapine reduces the chemokine-mediated migration, the expression of the chemokine receptors for CCL2 (CCR2) and CCL5 (CCR5) was analyzed on peripheral blood immune cells from healthy and EAE mice at day 7 after immunization treated with vehicle or clozapine commencing 1 day prior immunization; reflecting the first wave of infiltration into the CNS. Fig. 5 shows the expression of these two receptors on monocytes, neutrophils, and T cells (Fig. 5a–f) and reveals that overall, receptor expression was not altered in EAE animals compared to healthy control, except for a decrease in mean fluorescence intensity (MFI) for CCR2 on neutrophils in EAE compared to healthy control. However, no differences could be detected between vehicle or clozapine-treated mice following 7 days of EAE induction (Fig. 5). In addition, the frequency of CCR-positive cells within each cell type was also not altered by clozapine treatment (Additional file 4: Figure S4 a, b). From these results, we conclude that clozapine does not alter the expression of chemokine receptors directly to reduce migration but instead may alter factors downstream of these receptors.

CCR2 and CCR5 expression is similar between vehicle and clozapine treatment. C57BL/6 female mice were treated with clozapine or vehicle control in their drinking water for 7 days, blood was collected and analyzed by flow cytometry for CCR2 (a–c) and CCR5 (d–f) expression. Shown are the MFI and SEM of individual mice (n = 13/treatment group).*p < 0.0332, **p < 0.021, ***p < 0.0002, and ****p < 0.0001 by one-way ANOVA with Sidak’s multiple comparisons test
Clozapine inhibits migration by upregulation of cyclic AMP
All chemokine receptors are part of a family of G-protein coupled receptors and one of the consequences of chemokine receptor activation is a change in cAMP within the cell. High cAMP concentrations are associated with reduced cellular locomotion, adhesion, and migration (Iwasaki 1994; Laudanna 1997; ParmoCabanas 2004). Therefore, we assessed if cAMP levels changed in different immune cell populations after CCL2 and CCL5 treatment in the presence or absence of clozapine. As expected, stimulation for 18 h with forskolin, a labdane diterpene, led to elevated intracellular cAMP in cultured splenocytes compared to the vehicle treatment (Fig. 6a, b). When CCL5 (Fig. 6b) was added with forskolin, a significant reduction in cAMP concentration was observed compared to forskolin alone suggesting that high chemokine concentrations reduced intracellular cAMP concentration, which would increase migration. The addition of clozapine to splenocyte cultures containing both forskolin and CCL5 significantly increased the intracellular cAMP levels (Fig. 6b) suggesting reduced migration. However, no significant effect was detected with the addition of clozapine treatment to forskolin and CCL2 (Fig. 6a).

cAMP expression is altered by clozapine in splenocytes. Splenocytes (a, b) or sorted monocytes (c, d) were left untreated or stimulated with forskolin, CCL2 or CCL5 and clozapine in a single treatment or with different combinations and cAMP accumulation was measured with in the cells. Two technical replicates in three independent experiments per treatment group. *p < 0.0332, **p < 0.021, ***p < 0.0002 and by two-way ANOVA with Tukey’s multiple comparisons test
Because > 90% of splencocytes from healthy mice are B or T cells and these cells express lower levels of CCR2 (Additional file 4: Figure S4c, e), we repeated these experiments using isolated monocytes, which are one of the main cell types expressing CCR2. Monocytes were isolated from the spleen and stimulated with forskolin, the chemokines, and clozapine. Additionally, monocytes were stimulated with LPS to further increase cAMP concentrations, as in preliminary experiments only low levels of cAMP were detected without LPS stimulation (Additional file 4: Figure S4h). The addition of LPS did not change the expression of CCR2 on the cell surface of cultured monocytes (Additional file 4: Figure S4d, f). Forskolin stimulation in monocytes led to only a minor increase in cAMP levels and addition of the chemokines CCL2 or CCL5 did not alter the cAMP levels (Fig. 6c, d). While treatment with clozapine in addition to forskolin and CCL5 did not show an effect (Fig. 6d), treatment with clozapine in addition to forskolin and CCL2 unexpectedly reduced the cAMP levels compared to forskolin treatment or in combination with CCL2 and was no different to forskolin treatment alone (Fig. 6c). Overall, we found that clozapine inhibited CCL5-mediated downregulation of cAMP in splenocytes but unexpectedly decreased cAMP levels in CCL2 and forskolin-treated monocytes, and together, these findings indicate that clozapine can directly alter CCL5 and CCL2-mediated signal pathways in immune cells.
Discussion
The aim of this study was to investigate how the atypical antipsychotic agent, clozapine, reduces disease onset and severity in the EAE model of MS. EAE is characterized by a high infiltration of monocytes, neutrophils, and T cells into the CNS at the peak of disease [10, 25], and in this study, we found that clozapine reduced the infiltration of immune cells into the CNS and decreased the chemokine expression in the CNS in the early pre-symptomatic phase. In addition to reducing CCL2 and CCL5 expression, we found that clozapine treatment directly inhibited chemokine-mediated migration and signal pathways in immune cells. This study is the first to report a direct effect of clozapine on chemokine-induced signal pathways and reveals a novel mechanism by which clozapine may modulate immune responses.
In EAE, high chemokine production in the CNS is associated with acute disease symptoms [27], and chemokines, including MIP, CCL2, CCL3, CCL4, CCL5, CCL12, and CCL22, have been shown to play a role in development of EAE [28,29,30].
At the peak of disease, 14 days after immunization, high expression of the chemokines CCL2 and CCL5 were detected in the brain tissue of mice [13] and spinal cord [27]. Additionally, elevated CCL5 levels have been reported in the cerebrospinal fluid (CSF) of patients during MS relapses [31]. In contrast, CCL3 expression was not significantly changed at disease onset, although it is possible that it may play a role in later phase of EAE [13]. Here, the analysis of the early regulation of inflammatory chemokines showed that already by 7 days after immunization, an enhanced expression of CCL2 and CCL5 could be detected in brain tissue with the expression increasing by 11 days post-immunization in the spinal cord. This finding is in line with Borjini et al. where they showed increased chemokine expression in the onset phase of EAE in a rat model of multiple sclerosis. CCL2 was highly present in the cerebrospinal fluid (CSF) 8 days after EAE induction, while CCL5 was highest at 11 and 18 days after EAE induction [26]. This early expression of chemokine genes was also detected in the spinal cord 4 days after adoptively transferred T cells in the passive induction model of murine EAE [32]. In contrast to other chemokines of the C-C family, which trigger the Th1 phenotype upon their interaction with CCR5 on T-helper cells [33], CCL2 expression is associated with the polarization of Th0 cells toward a Th2 phenotype [34, 35]. This is characterized by an enhanced of IL-4 by T cells induced by CCL2 [36] and this early expression of CCL2 may also be associated with initiation of the blood-brain barrier breakdown leading to enhanced infiltration.
To investigate which cell types are the major source for CCL2 and CCL5 expression in the CNS and which cell types are targeted by clozapine treatment, primary microglia and astrocytes were isolated and bone marrow-derived macrophages were generated and treated with clozapine after activation. It has been shown that CCL2 is expressed by astrocytes and macrophages within actively demyelinating MS plaques [37] and astrocytes are known to be the likely cellular source of CCL2 in MS and EAE [38, 39]. Our results are in agreement with the previous published data showing that CCL2 and CCL5 are highly expressed after astrocyte activation; however, treatment with clozapine did not result in a decreased expression in astrocytes. However, CCL2 expression by astrocytes has been shown to be critical for the ongoing inflammation in chronic EAE and not for the induction phase [40]. Thus, CCL2 expression on other CNS resident cells may be important in the initial early inflammatory processes at disease onset.
Another major source of chemokine expression within the CNS is resident activated microglia. The production of CCL2 from resident microglia contributes to the recruitment of leukocytes into the CNS in EAE [41]. In our study, we could show that CCL2 and CCL5 mRNA was highly expressed after microglia activation and that either clozapine pre-treatment or co-treatment reduced the LPS-induced expression of CCL2 and CCLL5 mRNA. This reduction is likely due to reduced activation of microglia by clozapine as it has been shown previously by our group that clozapine reduces the expression of the activation markers I-A and CD40 on microglia in the brain and spinal cord during EAE [21]. In addition, clozapine was also shown to inhibit microglia activation in culture as measured by the reduced expression of Iba1 [42], and clozapine pre-treatment resulted in the suppression of LPS-induced expression of IL-1β, IL-6, and iNOS mRNA in both BV2 and primary cultured rat microglial cells [43]. We also found that bone marrow-derived macrophages are capable of expressing CCL2 and CCL5 after activation and that this expression was reduced with clozapine treatment. While in our case, it did not make a difference whether the macrophages were derived using M-CSF or GM-CSF, it has been shown previously that M-CSF-derived macrophages exhibit higher CCL2-dependent monocyte recruitment than GM-CSF-derived, whereas only the latter are capable of responding to CCL2 [44]. Overall, we show that microglia and macrophages are a major target of clozapine treatment leading to the reduced expression of the chemoattractant cytokines CCL2 and CCL5.
In addition to demonstrating a reduction in the signals driving migration into the CNS during EAE, we also found that clozapine directly inhibits the ability of these immune cells to migrate. Using an in-vitro migration assay of wound healing and an in-vivo CCL2 or CCL5-driven migration assay, we showed for the first time that migration was inhibited in the presence of clozapine. Our results suggest that clozapine may directly target the migrating immune cells and prevent their response to chemotactic signals like CCL2 and CCL5. The chemokine receptors CCR2 and CCR5 have been shown to be critically important in controlling leukocyte migration across endothelium and the blood-brain barrier [45] and to play an important role in the development and severity of EAE. CCR2−/− mice immunized with MOGp35–55 failed to develop mononuclear cell inflammatory infiltrates in the CNS and failed to increase CNS levels of the chemokines, MCP-1, and interferon (IFN)-inducible protein 10 (IP-10) as well as CCR1, CCR2, and CCR5 [46]. Additionally, deficiency in CCR5 suppresses EAE in C57BL/6 mice by reduced immune cells infiltration and astrocyte and microglia activation [47]. However, our work revealed that the block in migration was not mediated by changes in the expression of CCR2 and CCR5 on immune cells in the blood of EAE animals where we saw an overall reduction of CCR2 and CCR5 on neutrophils compared to healthy animals, but no difference with clozapine treatment. However, it was shown that CCR5 is present only on a small subset of circulating blood monocytes, but is highly increased on all monocytes in MS lesions [48]. In line with this, Trebst et al. showed that monocytes showed higher expression of CCR5 in the CNS than in the blood [49]. There is a good correlation between high expression of CCR2 and CCR5 in the spinal cord and disease severity and the main cellular source of CCR1, CCR2, and CCR5 high expression were migrating inflammatory cells [15, 45, 47]. So immune cells expressing higher levels of CCR2 or CCR5 in the blood could be migrating through the BBB into the CNS and accumulating in the lesions. This would explain why a reduction of CCR2 and CCR5 on immune cells in the blood are detected in our study. In addition, high levels present of the corresponding chemokines might lead to a faster and more frequent internalization of the existing chemokine receptors on the cell surface. Once the chemokine binds to its corresponding receptor, they are quickly internalized, and the downstream signaling is activated. CCR2 has a half-life on the cell surface of around 30 min, while CCR5 is internalized after around 60 min [50] but depending on the ligand, affinity, and cell type. This might, in addition, explain why in EAE where high levels of the chemokines are present, the receptors are downregulated.
Given that the chemokine receptors were not targeted by clozapine treatment, we speculated that the block in the response to chemotactic signals may be due to an alteration in the signaling cascade downstream of the receptors by clozapine. One signaling molecule that is associated with migration is the second messenger cAMP. cAMP accumulation is a representative read out for CCR5 activation, where all CCR5 agonists inhibited the cAMP production triggered by forskolin [51]. Treatment with clozapine in addition to forskolin and CCL5 reversed the CCL5-induced downregulation of cAMP. In contrast, only a minor difference was seen with CCL2 co-treatment and in addition with clozapine. This lack of response may be due to the higher expression of CCR5 on lymphocytes than CCR2. While monocytes express high levels of CCR2, we were unable to demonstrate a reproducible effect of clozapine on cAMP levels in monocytes. cAMP has many different functions in different cell types; it has the ability to inhibit proliferation in some cell types while stimulating proliferation in others. Thus, cAMP has cell type-specific effects demonstrating that the same signaling molecule can have opposing effects in the different cells [52]. In activated macrophages, cAMP plays a key role in the regulation of an inflammatory response, and an increase in cAMP reduces cytokine and chemokine production by activated macrophages leading to a dramatical dampening of the inflammatory response [53]. Inhibition of migration and locomotion of cells is usually associated with an elevation of intracellular cAMP; however, this is also concentration-dependent and cell type-specific, especially in epidermal cell migration [54]. For example, the presence of cAMP can reduce the number of cells bound to the adhesion molecule VCAM1, and PKA inhibitors counteract the decreased adhesion to VCAM1 induced by cAMP [55] suggesting that cAMP-dependent PKA acts as a negative modulator on the chemoattractant [56].
Forskolin is a highly potent and rapid inducer of cAMP. In line with our data, studies have shown that different chemokines exhibited the capacity to inhibit forskolin-induced cAMP accumulation in a dose-dependent manner. CCL5 but not CCL2 treatment reduced the forskolin-stimulated cAMP formation in a dose-dependent manner in HEK293 cells [57], and CCL5 also inhibited adenylyl cyclase activity in cells transiently transfected with CCR1 [58]. The reports on CCL2 by different groups are more complicated. While Wang et al. and O’Boyle et al. showed that interaction of CCL2 with CCR2 causes cAMP inhibition and a significant reduction in the concentration of cAMP [59, 60], Mizutani et al. showed that CCL2 increases cAMP accumulation and that the differences of CCL2-induced cAMP accumulation may indeed dependent on cell type [61]. It has also been shown that different ligands for the same receptor resulted in different outcomes. CCL2 and CCL8 revealed different maximal inhibition of forskolin-induced cAMP production by both chemokines suggesting that CCR2-mediated cAMP alteration is concentration dependent [62]. Taken together, these finding may explain why there is a difference between the response of lymphocyte and monocytes to CCL5 and CCL2. Finally, cAMP is not the only signaling molecule activated downstream of the chemokine receptors CCR5 and CCR2. It is known that CCL2 treatment increases Ca2+ flux and leads to PI3k-Akt activation [59, 61]. All these different pathways may influence the migratory potential of cells with each pathway having a different influence in different cell types and future work is needed to elaborate the effect of signaling pathway activation in the presence of clozapine.
Conclusion
In summary, this study is the first report of a novel mechanism of action for the atypical antipsychotic drug, clozapine, in EAE. Overall, we have shown that clozapine inhibited the migration of immune cells into the CNS in the EAE model of MS. This migration was prevented firstly by reduced chemokine expression in the CNS, and secondly by directly targeting the migratory potential of the cells. Out data suggests that clozapine did not alter the expression of the receptors directly but instead targeted the downstream signaling pathways that enable migration. Further studies need to elucidate in more detail the precise signaling pathways that are targeted and whether these activities are cell type-specific. Together, this work highlights a novel mechanism by which clozapine may exert the reported immunomodulatory effects that contribute to its known effectiveness in treating a wide range of neuroglial disorders including schizophrenia and Parkinson’s disease.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AC:
-
Adenylyl cyclases
- Akt:
-
Protein kinase B
- CCL:
-
Chemokine (C-C motif) ligand
- CCR:
-
C-C chemokine receptor
- CNS:
-
Central nervous systems
- CSF:
-
Cerebrospinal fluid
- EAE:
-
Experimental autoimmune encephalomyelitis
- ERK:
-
Extracellular signal-regulated kinases
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- IFNγ:
-
Interferon gamma
- IL:
-
Interleukin
- MCP-1:
-
Monocyte chemoattractant protein 1
- M-CSF:
-
Macrophage colony-stimulating factor
- MFI:
-
Mean fluorescence intensity
- MS:
-
Multiple sclerosis
- NOS:
-
Nitric oxide synthases
- PBS:
-
Phosphate-buffered saline
- PDE:
-
Phosphodiesterases
- PI3K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- RANTES:
-
Regulated upon activation normal T cell expressed and secreted
- TNFα:
-
Tumor necrosis factor alpha
- VEGF:
-
Vascular endothelial growth factor
References
Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. 2017;133(2):223–44.
Martin R, Lutterotti A. Molecular basis of multiple sclerosis; 2010.
Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230(4729):1043–5.
Methner A, Zipp F. Multiple sclerosis in 2012: novel therapeutic options and drug targets in MS. Nat Rev Neurol. 2013;9(2):72–3.
Hutchinson M. Natalizumab: a new treatment for relapsing remitting multiple sclerosis. Ther Clin Risk Manag. 2007;3(2):259–68.
Iarlori C, Reale M, De Luca G, Di Iorio A, Feliciani C, Tulli A, et al. Interferon beta-1b modulates MCP-1 expression and production in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2002;123(1–2):170–9.
Molnarfi N, Prod'homme T, Schulze-Topphoff U, Spencer CM, Weber MS, Patarroyo JC, et al. Glatiramer acetate treatment negatively regulates type I interferon signaling. Neurol Neuroimmunol Neuroinflamm. 2015;2(6):e179.
Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. 2011;1812(2):220–30.
Palmer AM. Multiple sclerosis and the blood-central nervous system barrier. Cardiovasc Psychiatry Neurol. 2013;2013:530356.
Stoolman JS, Duncker PC, Huber AK, Segal BM. Site-specific chemokine expression regulates central nervous system inflammation and determines clinical phenotype in autoimmune encephalomyelitis. J Immunol. 2014;193(2):564–70.
Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–21.
Mennicken F, Maki R, de Souza EB, Quirion R. Chemokines and chemokine receptors in the CNS: a possible role in neuroinflammation and patterning. Trends Pharmacol Sci. 1999;20(2):73–8.
dos Santos AC, Barsante MM, Arantes RM, Bernard CC, Teixeira MM, Carvalho-Tavares J. CCL2 and CCL5 mediate leukocyte adhesion in experimental autoimmune encephalomyelitis—an intravital microscopy study. J Neuroimmunol. 2005;162(1–2):122–9.
Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. 2005;25(5):593–606.
Glabinski AR, Bielecki B, O'Bryant S, Selmaj K, Ransohoff RM. Experimental autoimmune encephalomyelitis: CC chemokine receptor expression by trafficking cells. J Autoimmun. 2002;19(4):175–81.
Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, et al. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol. 1995;155(12):5769–76.
Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193(6):713–26.
Naheed M, Green B. Focus on clozapine. Curr Med Res Opin. 2001;17(3):223–9.
Meltzer HY, Alphs LD, Bastani B, Ramirez LF, Kwon K. Clinical efficacy of clozapine in the treatment of schizophrenia. Pharmacopsychiatry. 1991;24(2):44–5.
O'Sullivan D, Green L, Stone S, Zareie P, Kharkrang M, Fong D, et al. Treatment with the antipsychotic agent, risperidone, reduces disease severity in experimental autoimmune encephalomyelitis. PLoS One. 2014;9(8):e104430.
Green LK, Zareie P, Templeton N, Keyzers RA, Connor B, La Flamme AC. Enhanced disease reduction using clozapine, an atypical antipsychotic agent, and glatiramer acetate combination therapy in experimental autoimmune encephalomyelitis. Mult Scler J Exp Transl Clin. 2017;3(1):2055217317698724.
Zareie P, Connor B, La Flamme AC. Amelioration of experimental autoimmune encephalomyelitis by clozapine is not associated with defective CD4 T cell responses. J Neuroinflammation. 2017;14(1):68.
Stone S, La Flamme AC. Type II activation of macrophages and microglia by immune complexes enhances Th17 biasing in an IL-6-independent manner. PLoS One. 2016;11(10):e0164454.
Tierney JB, Kharkrang M, La Flamme AC. Type II-activated macrophages suppress the development of experimental autoimmune encephalomyelitis. Immunol Cell Biol. 2009;87(3):235–40.
Barthelmes J, Tafferner N, Kurz J, de Bruin N, Parnham MJ, Geisslinger G, et al. Induction of Experimental Autoimmune Encephalomyelitis in Mice and Evaluation of the Disease-dependent Distribution of Immune Cells in Various Tissues. J Vis Exp. 2016;e53933(111):1–10.
Borjini N, Fernandez M, Giardino L, Calza L. Cytokine and chemokine alterations in tissue, CSF, and plasma in early presymptomatic phase of experimental allergic encephalomyelitis (EAE), in a rat model of multiple sclerosis. J Neuroinflammation. 2016;13(1):291.
Fischer FR, Santambrogio L, Luo Y, Berman MA, Hancock WW, Dorf ME. Modulation of experimental autoimmune encephalomyelitis: effect of altered peptide ligand on chemokine and chemokine receptor expression. J Neuroimmunol. 2000;110(1–2):195–208.
Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14(11):1189–97.
Forde EA, Dogan RN, Karpus WJ. CCR4 contributes to the pathogenesis of experimental autoimmune encephalomyelitis by regulating inflammatory macrophage function. J Neuroimmunol. 2011;236(1–2):17–26.
Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. 1995;155(10):5003–10.
Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103(6):807–15.
Godiska R, Chantry D, Dietsch GN, Gray PW. Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol. 1995;58(2):167–76.
Van Coillie E, Van Damme J, Opdenakker G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev. 1999;10(1):61–86.
Chensue SW, Warmington KS, Lukacs NW, Lincoln PM, Burdick MD, Strieter RM, et al. Monocyte chemotactic protein expression during schistosome egg granuloma formation. Sequence of production, localization, contribution, and regulation. Am J Pathol. 1995;146(1):130–8.
Handel TM, Domaille PJ. Heteronuclear (1H, 13C, 15N) NMR assignments and solution structure of the monocyte chemoattractant protein-1 (MCP-1) dimer. Biochemistry. 1996;35(21):6569–84.
Karpus WJ, Ransohoff RM. Cutting edge commentary: chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. J Immunol. 1998;161(6):2667–71.
Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other β-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84(2):238–49.
Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, et al. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7(6):592–600.
Hulkower K, Brosnan CF, Aquino DA, Cammer W, Kulshrestha S, Guida MP, et al. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J Immunol. 1993;150(6):2525–33.
Kim RY, Hoffman AS, Itoh N, Ao Y, Spence R, Sofroniew MV, et al. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J Neuroimmunol. 2014;274(1–2):53–61.
Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J Immunol. 2008;180(11):7376–84.
Hu X, Zhou H, Zhang D, Yang S, Qian L, Wu HM, et al. Clozapine protects dopaminergic neurons from inflammation-induced damage by inhibiting microglial overactivation. J NeuroImmune Pharmacol. 2012;7(1):187–201.
Jeon S, Kim SH, Shin SY, Lee YH. Clozapine reduces toll-like receptor 4/NF-kappaB-mediated inflammatory responses through inhibition of calcium/calmodulin-dependent Akt activation in microglia. Prog Neuro-Psychopharmacol Biol Psychiatry. 2018;81:477–87.
Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192(8):3858–67.
Simpson J, Rezaie P, Newcombe J, Cuzner ML, Male D, Woodroofe MN. Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol. 2000;108(1–2):192–200.
Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192(7):1075–80.
Gu SM, Park MH, Yun HM, Han SB, Oh KW, Son DJ, et al. CCR5 knockout suppresses experimental autoimmune encephalomyelitis in C57BL/6 mice. Oncotarget. 2016;7(13):15382–93.
Ubogu EE, Cossoy MB, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27(1):48–55.
Trebst C, Sorensen TL, Kivisakk P, Cathcart MK, Hesselgesser J, Horuk R, et al. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am J Pathol. 2001;159(5):1701–10.
Neel NF, Schutyser E, Sai J, Fan GH, Richmond A. Chemokine receptor internalization and intracellular trafficking. Cytokine Growth Factor Rev. 2005;16(6):637–58.
Corbisier J, Gales C, Huszagh A, Parmentier M, Springael JY. Biased signaling at chemokine receptors. J Biol Chem. 2015;290(15):9542–54.
Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie lecture delivered on 5 July 2003 at the special FEBS meeting in Brussels. FEBS J. 2005;272(14):3491–504.
Hertz AL, Bender AT, Smith KC, Gilchrist M, Amieux PS, Aderem A, et al. Elevated cyclic AMP and PDE4 inhibition induce chemokine expression in human monocyte-derived macrophages. Proc Natl Acad Sci U S A. 2009;106(51):21978–83.
Iwasaki T, Chen JD, Kim JP, Wynn KC, Woodley DT. Dibutyryl cyclic AMP modulates keratinocyte migration without alteration of integrin expression. J Invest Dermatol. 1994;102(6):891–7.
Parmo-Cabanas M, Bartolome RA, Wright N, Hidalgo A, Drager AM, Teixido J. Integrin alpha4beta1 involvement in stromal cell-derived factor-1alpha-promoted myeloma cell transendothelial migration and adhesion: role of cAMP and the actin cytoskeleton in adhesion. Exp Cell Res. 2004;294(2):571–80.
Laudanna C, Campbell JJ, Butcher EC. Elevation of intracellular cAMP inhibits RhoA activation and integrin-dependent leukocyte adhesion induced by chemoattractants. J Biol Chem. 1997;272(39):24141–4.
Aramori I, Zhang J, Ferguson SSG, Bieniasz PD, Cullen BR, Caron MG. Molecular mechanism of desensitization of the chemokine receptor Ccr-5 : receptor signaling and internalization are dissociable from its role as an Hiv-1 Co-receptor. EMBO J. 1997;16(15):4606–16.
Tian Y, New DC, Yung LY, Allen RA, Slocombe PM, Twomey BM, et al. Differential chemokine activation of CC chemokine receptor 1-regulated pathways: ligand selective activation of Galpha 14-coupled pathways. Eur J Immunol. 2004;34(3):785–95.
Wang J, Zhuang ZG, Xu SF, He Q, Shao YG, Ji M, et al. Expression of CCL2 is significantly different in five breast cancer genotypes and predicts patient outcome. Int J Clin Exp Med. 2015;8(9):15684–91.
O'Boyle G, Brain JG, Kirby JA, Ali S. Chemokine-mediated inflammation: identification of a possible regulatory role for CCR2. Mol Immunol. 2007;44(8):1944–53.
Mizutani K, Roca H, Varsos Z, Pienta KJ. Possible mechanism of CCL2-induced Akt activation in prostate cancer cells. Anticancer Res. 2009;29(8):3109–13.
Berchiche YA, Gravel S, Pelletier ME, St-Onge G, Heveker N. Different effects of the different natural CC chemokine receptor 2b ligands on beta-arrestin recruitment, Galphai signaling, and receptor internalization. Mol Pharmacol. 2011;79(3):488–98.
Acknowledgments
The authors would like to thank Pirooz Zareie and Sven Sondhauss for her help and technical advice and Matt Rowe for the primary astrocytes.
Funding
This study was funded by the Ministry of Business, Innovation, and Employment.
Author information
Authors and Affiliations
Contributions
KR performed most of the experiments, analyzed the data, and wrote the manuscript. VP performed the in-vitro migration experiments and analyzed them. BC contributed to the experimental design. ACL contributed to the experimental design, data analysis and edited the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Victoria University of Wellington Animal Ethics Committee approved all experimental procedures used in this study (2014-R23).
Consent for publication
Not applicable.
Competing interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Anne Camille La Flamme and Bronwen Connor have a patent for the use of clozapine during MS. The authors declare no other financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Figure S1. Clozapine treatment reduces disease severity at the onset of EAE. C57BL/6 female mice were treated with clozapine (60 mg/kg/day) or vehicle control in their drinking water commencing one day prior to immunization and were scored and weight daily. At day 5, 7, 9 and 11 after EAE induction spinal cord (a), brain (b), spleen (c) and blood (d) was collected, cells were isolated and counted. Shown are the means and SEM of individual mice (n = 9/treatment group) from three independent experiments normalized to healthy vehicle for each day. (e) C57BL/6 female mice were treated with clozapine or vehicle control in their drinking water. At day 5, 7, 9 and 11 after EAE induction brain was collected, lysed and protein expression was analyzed by Milliplex. Shown are the means and SEM of individual mice (n = 3/ treatment group) normalized to healthy vehicle for each day.
Additional file 2:
Figure S2. Clozapine treatment shows a minor effect on the cell numbers and CCL2 and CCL5 expression in spleen or blood. C57BL/6 female mice were treated with clozapine (60 mg/kg/day) or vehicle control in their drinking water commencing one day prior to immunization and were scored daily. At day 5, 7, 9 and 11 after EAE induction spleen (a,b) and blood (c,d) was collected and RNA was extracted and analyzed by qRT-PCR for CCL2 and CCL5 expression. Shown are the means and SEM of individual mice (n = 9/ treatment group) from 3 independent experiments normalized to cyclophilin A as a housekeeper and healthy vehicle for each day.
Additional file 3:
Figure S3. Clozapine treatment reduces CCL2- and CCL5-mediated migration. (a) Representative images of BMMO in the in-vitro migration assay (b-k) C57BL/6 female mice were treated with clozapine (60 mg/kg/day) or vehicle control in their drinking water for 7 days. At the last day, CCL2 or CCL5 (left hind flank) or PBS (right hind flank) was injected s.c., and 18 h later, the draining LN cells were isolated and analyzed by flow cytometry. The total number of the individual cell types after CCL2 or CCL5 injection in the LN from 3 independent experiments (n = 13–14/group) are shown. P – PBS, 2 – CCL2, 5 – CCL5, *p < 0.0332 by paired 1-way ANOVA with Sidak’s multiple comparisons test.
Additional file 4:
Figure S4. CCR2 and CCR5 expression is unaltered. C57BL/6 female mice were treated with clozapine (60 mg/kg/day) or vehicle control in their drinking water commencing one day prior to immunization and were scored daily. At day 5, 7, 9 and 11 after EAE induction blood was collected analyzed by flow cytometry for CCR2 (a) or CCR5 (b) expression. Shown are the frequency of the parent population and SEM of individual mice (n = 13/ treatment group). *p < 0.0332, **p < 0.021 and ***p < 0.0001 by 1-way ANOVA with Sidak’s multiple comparisons test. CCR2 and CCR5 expression on splenocytes (c,e) and sorted monocyte (d,f) after culture, representative flow plots and dMFI of receptor to isotype control. (g) cAMP measurement in splenocytes treated with forskolin and ConA. (h) cAMP measurement in sorted monocytes treated with forskolin and LPS. p < 0.0332, **p < 0.021 and by 2-way ANOVA with Tukey’s multiple comparisons test.
Additional file 5:
Figure S5. Gating strategy for infiltration and in-vivo migration experiments. Gating strategy is shown for the (a) infiltration experiments for brain/spinal cord and spleen/blood (Fig. 1 c-r) and (b) in-vivo migration experiment for lymph nodes (Fig. 4 b-i and Additional file 3 b-k) from one EAE vehicle treated animal as an example, the same flow cytometry markers are used for both experiments: CD4-BV521 (RM4–5), CD45-BV510 (30-F11), CD25-AF488 (PC61), CD8-PerCPCy5.5 (53–6.7), CD11b-PE-Cy7 (M1/70), CD3-APC-Cy7 (17A2), Ly6C-PE (HK1.4), Ly6G-APC (1A8).
Additional file 6:
Figure S6. Gating strategy for CCR expression in blood. Gating strategy is shown for CCR expression analysis in blood (Fig. 5 and Additional file 4 a,b) from one EAE vehicle treated animal as an example, the following antibodies were used to detect the populations: CD4-BV521 (RM4–5), CD45-BV510 (30-F11), CD8-PerCPCy5.5 (53–6.7), CD11b-PE-Cy7 (M1/70), CD45R-AF488 (RA3-6B2), CCR2-PE (475301), CCR5-APC (HM-CCR5) Gr1-APC-Cy7 (RB6-8C5).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Robichon, K., Patel, V., Connor, B. et al. Clozapine reduces infiltration into the CNS by targeting migration in experimental autoimmune encephalomyelitis. J Neuroinflammation 17, 53 (2020). https://doi.org/10.1186/s12974-020-01733-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-020-01733-4