
Abstract
Background
ABCA4, the gene implicated in Stargardt disease (STGD1), contains 50 exons, of which 17 contain multiples of three nucleotides. The impact of in-frame exon skipping is yet to be determined. Antisense oligonucleotides (AONs) have been investigated in Usher syndrome-associated genes to induce skipping of in-frame exons carrying severe variants and mitigate their disease-linked effect. Upon the identification of a STGD1 proband carrying a novel exon 17 canonical splice site variant, the activity of ABCA4 lacking 22 amino acids encoded by exon 17 was examined, followed by design of AONs able to induce exon 17 skipping.
Methods
A STGD1 proband was compound heterozygous for the splice variant c.2653+1G>A, that was predicted to result in in-frame skipping of exon 17, and a null variant [c.735T>G, p.(Tyr245*)]. Clinical characteristics of this proband were studied using multi-modal imaging and complete ophthalmological examination. The aberrant splicing of c.2653+1G>A was investigated in vitro in HEK293T cells with wild-type and mutant midigenes. The residual activity of the mutant ABCA4 protein lacking Asp864-Gly885 encoded by exon 17 was analyzed with all-trans-retinal-activated ATPase activity assay, along with its subcellular localization. To induce exon 17 skipping, the effect of 40 AONs was examined in vitro in WT WERI-Rb-1 cells and 3D human retinal organoids.
Results
Late onset STGD1 in the proband suggests that c.2653+1G>A does not have a fully deleterious effect. The in vitro splice assay confirmed that this variant leads to ABCA4 transcripts without exon 17. ABCA4 Asp864_Gly863del was stable and retained 58% all-trans-retinal-activated ATPase activity compared to WT ABCA4. This sequence is located in an unstructured linker region between transmembrane domain 6 and nucleotide-binding domain-1 of ABCA4. AONs were designed to possibly reduce pathogenicity of severe variants harbored in exon 17. The best AON achieved 59% of exon 17 skipping in retinal organoids.
Conclusions
Exon 17 deletion in ABCA4 does not result in the absence of protein activity and does not cause a severe STGD1 phenotype when in trans with a null allele. By applying AONs, the effect of severe variants in exon 17 can potentially be ameliorated by exon skipping, thus generating partial ABCA4 activity in STGD1 patients.
Graphical abstract

Similar content being viewed by others


Introduction
Biallelic variants in the ABCA4 gene are implicated in Stargardt disease type 1 (STGD1) [1], characterized by progressive macular degeneration, fundus flecks caused by amassed toxic metabolites formed during the visual cycle and a peripapillary region resistant to disease-associated changes [2]. Onset can be in the first decade of life which has been described as a panretinal cone-rod dystrophy [3, 4]. More recently, a better appreciation of the role of mild or hypomorphic variants, when in trans with severe or null alleles, has revealed an increasing number of persons with late-onset STGD1 [5,6,7]. STGD1 is considered the most prevalent form of inherited macular dystrophy and it is estimated that there are 1,385,000 affected individuals worldwide [8]. Interestingly, some of the mild variants have been shown, or are suspected, to be incompletely penetrant, when in trans with a severe or null allele. In addition, a sex imbalance (more females than males affected) was observed for probands carrying c.5603A>T, p.(Asn1868Ile) or c.5882G>A, p.(Gly1961Glu), in trans with a severe or null allele [5, 6]. These results suggest that a significant part of STGD1 probands are subject to polygenic or multifactorial inheritance in which biallelic ABCA4 variants are needed but not enough to develop STGD1.
The structure of ABCA4 was reported to consist of two transmembrane domains (TMD), each consisting of 6 helical membrane spanning segments, two glycosylated exocytoplasmic domains (ECD) and two nucleotide-binding domains (NBD) divided over two non-symmetric consecutive halves [9, 10]. The cryo-EM structure of the 2273 amino acid transporter has been previously reported in its substrate-bound, nucleotide-bound and unbound states [11,12,13]. These studies gave insight in its size and suggested that the ABCA4 localization in the rims of photoreceptors’ outer segment disk membranes is predetermined because of its elongated ECDs which do not fit in the flattened parts of the photoreceptor disks. ABCA4 plays a critical role in the visual cycle by acting as a flippase for retinoids, which limits the accumulation of toxic bisretinoid compounds responsible for retinal degeneration [14].
To date, STGD1 remains an untreatable condition. Even though several clinical trials are ongoing (NCT03033108, NCT05244304, NCT03364153, NCT04545736), none of the investigated therapeutic approaches tackles the molecular genetic defects underlying the disease but is rather focused on managing the toxic vitamin-A metabolites or suppressing the complement component 5 in the complement system to reduce inflammation in the retinal pigment epithelium upon accumulation of A2E and bisretinoids [15]. Based on the variants’ allele numbers reported by Cornelis et al. in biallelic probands [16], 19% of 2167 unique variants and 23% of all 11,146 disease-associated alleles, affect splicing. The high incidence of splicing variants, of which many result in the insertion of pseudoexons in the mRNA, make STGD1 a great target for antisense oligonucleotide (AON)-based therapies.
Moreover, AONs can be used to redirect the splicing and skip coding regions without interfering with the translation of the rest of the protein [17]. AONs were firstly used to suppress expression of several disease-causing genes [18,19,20,21,22], with formivirsen pioneering as the first FDA-approved AON for treatment of cytomegalovirus retinitis [23]. Splicing modulating AON-based therapies were introduced in 2016 with Sarepta Therapeutics’ eteplirsen and, later on, golodirsen, which excluded ‘in-frame’ exons 51 or 53 in DMD, respectively. The open reading frame was maintained and the severe phenotype of Duchenne muscular dystrophy (DMD) due to protein-truncating variants in these exons was ameliorated [24, 25].
AON-based skipping of a coding region was also previously investigated for USH2A-associated retinitis pigmentosa (RP) and Usher syndrome (USH) variants that result in a premature stop of translation or in damaging amino acid substitutions [26,27,28]. The clinical trials focused on probands carrying the frequent USH2A exon 13 variants c.2299delG, p.(Glu767Serfs*21) and c.2276G>T, p.(Cys759Phe) [26].
In ABCA4, AONs were applied to either exclude pathogenic intronic insertions (a.k.a. pseudoexons) or restore wild-type splicing of exons 39 and 40 due to c.5461-10T>C [29,30,31,32,33,34,35]. Therapeutic intervention through the exclusion of exons carrying pathogenic variants was not yet considered as the above-described structure of the protein suggested that the complete coding region was needed for a correct function of ABCA4. ABCA4, consisting of 50 protein-coding exons, contains 17 exons consisting of a multiple of three nucleotides, which, if skipped, hypothetically, would not disrupt the open reading frame and a shorter yet potentially functional protein would be translated. In-frame skipping of nonessential exons could be a therapeutic approach for alleviating the effect of severe missense variants in ABCA4, however, the effect of in-frame exon skipping is unknown.
In this study, we report the first evidence of in-frame exon skipping that results in significant activity of ABCA4. We identified that the 22 amino acids encoded by exon 17, which are located entirely in a segment connecting TMD1 and NBD1 in ABCA4, are not crucial for the protein’s function. Following these findings, we developed an AON-based intervention designed to skip exon 17 that could potentially be used to treat severe variants harbored in this exon and increase the residual activity of ABCA4 in STGD1 patients.
Materials and methods
Clinical analysis
Clinical examination was performed with standard slit lamp and indirect ophthalmoscopy. Visual acuity was measured using ETDR charts. Fundus autofluorescence (FAF), spectral domain optical coherence tomography (SD-OCT) and fluorescein angiography (FA) were performed using Spectralis (HRA, Heidelberg Engineering, Heidelberg, Germany). Full field electroretinography (ffERG) was performed using a Ganzfeld bowl with standard ISCEV protocol (MonPackOne, Metrovision, Perenchies, France). Normative population data was used for comparison of the results for electrophysiology.
Construction and characterization of the ABCA4 c.2653+1G>A midigene
The BA13 construct spanning ABCA4 exons 16, 17 and 18, and 591 bp of upstream and 1211 bp of downstream intronic sequences, was used as WT template for generating the ABCA4 c.2653+1G> A midigene [36]. The variant was inserted by mutagenesis using the following primer sequences: 5′-ttggcttggcggtgaagatgagtcctttaaaacac-3′ and 5′-gtgttttaaaggactcatcttcaccgccaagccaa-3′. The mutated ABCA4 sequence was placed in the pCI NEO vector [37] by the Gateway cloning system as described previously [38]. The construct was linearized with SalI (New England Biolabs, Ipswich, MA, USA) and its sequence was validated with PacBio sequencing.
HEK293T cells were transfected with the mutant and WT constructs in 12-well plates at a concentration of 100 ng/well. The cells were harvested 48 h later and their RNA was extracted, and reverse-transcribed in cDNA as reported elsewhere [38]. To assess the isoform content of the cDNA, the forward primer binding in ABCA4 exon 16 (5′-agatggaagccatgttggag-3′) and the reverse primer annealing to exon 18 (5′-tggaatctcttgaccagcag-3′) were used in RT-PCR as described previously [36]. The sequences of the identified isoforms were determined with Sanger sequencing using the above-mentioned forward primer in ABCA4 exon 16.
Construction, localization and functionality assessment of ABCA4 ΔAsp864_Gly885
The WT ABCA4 coding sequence was introduced in the pcDNA™3.1 ( + ) Mammalian Expression Vector (GenScript, Nanjing, China). To exclude exon 17, the wild-type plasmid and a gBlock sequence (Integrated DNA Technologies, Coralville, IA, USA) lacking exon 17 were digested with 10U of Acc65I (Thermo Fisher Scientific, Waltham, MA, USA) and FseI (New England Biolabs, Ipswich, MA, USA). The two fragments were ligated and transformed in GT115 competent cells (InvivoGen, San Diego, CA, USA) as described previously [31].
The expression and purification of WT and ABCA4 variants were carried out as previously described [39]. Briefly, HEK293T cells were transfected with plasmid DNA (5 µg/10 cm plate) using 1 mg/ml polyethylenimine (PEI) MAX (Polysciences, Warrington, PA, USA) at a ratio PEI(µl):DNA(µg) 3:1. Cells were grown in a humidified incubator (5% CO2) at 37 °C in Dulbecco’s modified Eagle’s medium supplemented with 8% bovine growth serum, 200-µM l-glutamine and 1X antibiotic–antimycotic (Thermo Fisher Scientific, Waltham, MA, USA). After 24 h, cells were solubilized in buffer A (25 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid, pH 7.4, 0.15 mg/ml brain polar lipid (BPL), and 0.07 mg/ml 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 150 mM NaCl, 1 mM MgCl2, 10% glycerol, and 1 mM dithiothreitol) containing 18 mM CHAPS detergent and protease inhibitor cocktail by slowly adding one plate of cells resuspended in 100 µl of buffer A to 0.5 ml solubilization buffer. The solution was stirred for 30 min at 4 °C and subsequently centrifuged for 10 min at 100,000g in a Beckman Optima TL centrifuge using a TLA110.4 rotor. The supernatants from cell lysates were mixed with 50 µl of Rim-3F4 Sepharose 2B immunoaffinity matrix for 1 h at 4 °C. The matrix was washed six times in buffer A containing 10 mM CHAPS. The ABCA4 protein was eluted two times with 60 µl each in the same buffer containing 0.5 mg/ml 3F4 peptide at 18 °C for 60 min.
An aliquot was analyzed on Coomassie blue-stained SDS gels for quantification of protein expression. The remaining sample was directly used for ATPase activity assays using the ADP–Glo Kinase Assay luminescence detection protocol (Promega, Madison, WI, USA). Briefly, a 20-µl ATPase reaction was performed by incubating 20–40 ng of protein with either 1 µl of 0.8 mM all-trans retinal (ATR) or buffer for 15 min followed by the addition of 4 µl of 2.5 mM ultrapure ATP for 40 min at 37 °C. Each reaction was done in triplicate. Six microliters of each reaction were then added to a well of a 384 Corning white polystyrene microtiter plate, followed by 6 µl of ADP–Glo reagent and 12 µl of kinase detection reagent. The luminescence signal was detected using on a Molecular Device SpectraMax M3 spectrometer equipped with SoftMax Pro 5.4 software. The ATP-deficient mutant ABCA4-MM in which the lysine residues in the Walker A motif of NBD1 and NBD2 were substituted for methionine, p.(Lys969Met), and p.(Lys1978Met), respectively, was used to subtract background luminescence [40].
For the localization study, HEK293T cells were grown on polylysine treated glass coverslips and transfected as described above. After 24 h, cells were fixed in 2% paraformaldehyde/0.lM phosphate buffer, pH 7.2 for 15 min and washed with 0.lM phosphate buffer 3 × 15 min.
Cell membranes were blocked and permeabilized with 10% normal goat serum, phosphate buffer and 0.1% Triton X-100 for 30 min. ABCA4 was detected using monoclonal RIM-3F4 and Alexa Fluor 568 anti-mouse secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA) and Calnexin using polyclonal anti-calnexin (Abcam, Cambridge, UK) and Alexa Fluor 488 anti-rabbit secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA) in phosphate buffer containing 0.3% normal goat serum and 0.075% Triton X-100. Fluorescence was detected using a Zeiss LSM700 confocal microscope (Carl Zeiss, Oberkochen, Germany).
ARPE-19 (ATCC, Manassas, VA, USA) cells were cultured in Advanced DMEM/F-12 medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10% Fetal Bovine Serum (Biowest, Nuaillé, France) at 37 °C with 5% CO2. At 80–90% confluency, the cells were harvested and 3 × 104 cells were seeded in a µ-Slide 8 Well chambered coverslip (ibidi GmbH, Gräfelfing, Germany). The cells were transfected 24 h later with 250 ng of mutant or wild-type plasmid and incubated at 37 °C for additional 48 h. Part of the cells were used for immunoblotting for quantification of protein expression by following the previously described protocol [31], and part was used for immunostaining. For the latter, the cell cultures were rinsed with ice-cold PBS (Thermo Fisher Scientific, Waltham, MA, USA) with 5% Blocker BSA (Thermo Fisher Scientific, Waltham, MA, USA), fixed in cold 4% paraformaldehyde (Thermo Fisher Scientific, Waltham, MA, USA) and incubated at RT for 20 min. Next, these were permeabilized with ice-cold PBS + 0.1% Triton X-100 (Promega, Madison, WI, USA) for 10 min and blocked with PBS + 5% BSA for 1 h at 4 °C. The cells were incubated with anti-Calnexin antibody (1:200, Thermo Fisher Scientific, Waltham, MA, USA) and anti-ABCA4 Antibody, clone 3F4 (1:100, Abcam, Cambridge, UK) overnight at 4 °C. The nuclei were stained with Hoechst 33342 (1:1000, Thermo Fisher Scientific, Waltham, MA, USA). The images were collected with an LSM 800 confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 20 × objective and analyzed with ZEN Blue edition (Carl Zeiss, Oberkochen, Germany).
Antisense oligonucleotide design and screening in wild-type WERI-Rb-1 cells
The AONs were designed as described previously by Kaltak et al. [31] as an oligo-walk in order to target the ABCA4 exon 17 and the adjacent 60 nt and 16 nt of introns 16 and 17, respectively.
WERI-Rb-1 cells were grown as grape-like clusters in suspension in culture flasks with Roswell Park Memorial Institute 1640 Media (RPMI 1640, Thermo Fisher Scientific, Waltham, MA, USA) enriched with 10% Fetal Bovine Serum (FBS, Biowest, Nuaillé, France) at 37 °C with 5% CO2.
5 × 105 cells were transferred in 12-well plates and the AONs (ProQR Therapeutics, Leiden, the Netherlands), reported in Additional file 1: Table S1, were added to the cell culture medium at 10 µM as described previously [26]. The RNA was extracted, reverse-transcribed into cDNA and the isoform content was investigated using digital PCR (dPCR) by following the previously reported protocol [31]. The measurement of total ABCA4 was conducted in the latter by employing an assay consisting of primers that bound to exons 8 and 9, along with a probe targeting the junction of these two exons. Similarly, ABCA4 Δexon 17 was assessed using an assay that comprised primers binding in exons 16 and 18, and a probe targeting the junction of these two exons. The sequences of the assays are reported in Additional file 1: Table S2.
Generation of wild-type retinal organoids
StemRNA Human induced pluripotent stem cell line (iPSC) 771-3G (ReproCell, Yokohama, Japan) was cultured in 6-well plates coated with Corning® Matrigel® hESC-Qualified Matrix (Corning, NY, USA) containing 2 ml of mTeSR™1 medium (Stemcell Technologies, Vancouver, Canada) enriched with 25% mTeSR™1 5X Supplement (Stemcell Technologies, Vancouver, Canada) and 1% Penicillin/streptomycin (Sigma-Aldrich, Burlington, MA, USA). The iPSCs were differentiated in ROs by applying the protocol described by Hallam et al. [41].
Antisense oligonucleotide treatment in wild-type retinal organoids
AONs were administered to 190-days-old ROs at a 10 µM concentration; the culture medium was fully removed and fresh medium with AON was added. Every two days half of the culture medium was removed and replaced by fresh culture medium; this “wash-out” regimen was followed for 4 weeks. Next, the culture medium was removed completely, the ROs were washed in PBS and harvested in 300 µl of TRIreagent (Zymo Research, Irvine, CA, USA). The samples were snap frozen in liquid nitrogen and stored at − 80 °C. The ROs were lysed with a 25-gauge needle, and the RNA was extracted with the Direct-Zol RNA MicroPrep kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. 100 ng of extracted RNA was reverse transcribed in cDNA as described above. The samples were analyzed with isoform-specific assays for total ABCA4 and ABCA4 Δexon 17 (Additional file 1: Table S2) as described above.
To ensure the quality of ROs, the levels of retinal markers CRX, OPN1MW, and RHO were utilized, with a minimum threshold of more than 1000 copies/ng RNA. Samples that fell below all three thresholds were not included in the analysis. The positive and negative partitions were distinguished based on manually set thresholds in all experiments. To adjust for variations in cDNA input, the identified ABCA4 isoforms were normalized to the geometric mean of CRX, RHO, and OPN1MW.
The expression of additional retinal markers, i.e. NR2E3 (Hs00183915_m1, Thermo Fisher Scientific, Waltham, MA, USA) and NRL (Hs00172997_m1, Thermo Fisher Scientific, Waltham, MA, USA), was assessed to assure the integrity of marker expression before and after AON treatment.
Transcript data analysis
Using the QIAcuity Software Suite (Qiagen, Hilden, Germany), the positive and negative populations were separated with manually set thresholds, and the positive population was considered for further calculations. The formula used to calculate the percentage of AON-induced ABCA4 exon 17 skip in WERI-Rb-1 cells and ROs was the following: % ABCA4 Δexon 17 relative to total ABCA4 = ABCA4 Δexon 17/ABCA4 exons 8-9.
In silico off-target assessment for the best performing antisense oligonucleotides
To determine the potential off-target effects of AONs used in the treatment administered to ROs, an in silico analysis on human genomic DNA and transcriptome described previously was performed. Homo sapiens DNA (RefSeq_Gene) and mRNA (refseq_rna) sequence databases were investigated manually with the Basic Local Alignment Search Tool (https://blast.ncbi.nlm.nih.gov, accension date 15 May 2023).
Statistical analysis
All graphs represent data as mean ± SEM. The mean percentages of the AON efficiency and expression of retinal markers were analyzed for statistical significance vs. untreated or scrambled AON using GraphPad Prism 9 with ordinary one-way ANOVA test followed by Dunnett’s multiple comparison test. The mean numbers of CSS variants nearby multiple-of-three exons and non multiple-of-three-exons were investigated for statistical significance with an unpaired two-tailed t test with Welch’s correction. P ≤ 0.05 was considered statistically significant.
Results
Ophthalmogenetic findings in a STGD1 proband carrying ABCA4 c.2653+1G>A and a null allele
We identified a French STGD1 proband that carried a previously reported null variant c.735T>G, p.(Tyr245*) [42] and a novel canonical splice site variant c.2653+1G>A. Segregation analysis in the mother and brother revealed that these two variants were in trans. Interestingly, this proband presented with late onset symptoms (at 43 years of age), but consulted a ophthalmologist just 6 years later, in 2010, when fundus abnormalities were detected. The initial best corrected visual acuity (BCVA) measured in Monoyer scale was 10/10 for both eyes (Snellen equivalent 20/20). At the time, the main reported complaints were hemeralopia and photophobia. The initial exams revealed normal anterior segments on both eyes at slit lamp examination and no major refractive issues. Fundus examination revealed the appearance of a flecked retina with obvious signs of atrophy, as shown in Fig. 1A. Fundus autofluorescence (FAF) identified hyperautofluorescent lesions localized at the macula and extending about one disc diameter outside the vascular limits of the macula (Fig. 1B). Extensive central retinal atrophy was also noted. The presence of foveal sparing was confirmed by spectral domain optical coherence tomography (SD-OCT) (Fig. 1C). FAF also revealed peripapillary sparing. Fluorescein angiography (FA) showed signs of choroidal silence in the very early phases, more evident in the fleck and atrophy free zones extending beyond the mid periphery (Fig. 1D). Full field electroretinography (ffERG) showed subnormal scotopic and photopic responses for both eyes compared to the normative data for our center (Additional file 1: Fig. S1). BCVA remained at 20/20 for five years, then declined progressively and symmetrically over the next few years diminishing to 20/25 on 2016. In 2019, BCVA further decreased to 20/50 on both eyes and has been stable since. This is secondary to the foveal sparing seen up until his last exam on SD-OCT (Fig. 1E–P).

Multimodal imaging of both eyes of the proband. A–D Analysis at baseline performed at proband’s age of 49 years. (OD, oculus dexter or right eye; OS, oculus sinister or left eye) (A′, A″) Color fundus photographs reveal extensive macular atrophy with some pigment clumps more noticeable on the right eye along the border of the atrophy. There is some retro foveal retinal tissue remaining. B′, B″ Fundus autofluorescence (FAF) images where a large amount of hyperautofluorescent coalescent flecks is evident extending outside the vascular arcades but remaining within the posterior pole. The foveal sparing is greater on the right eye. Peripapillary sparing is present in both eyes. C′, C″ Spectral domain optical coherence tomography (SD-OCT) scan images passing through the fovea, shows clumping of the retinal pigment epithelium and extensive atrophy of the external retinal layers with retro foveal sparing. D′, D″ Fluorescein angiography mid phase images of the posterior pole. A window defect is present on both eyes in the areas presenting atrophy and beyond the posterior pole choroidal silence is present. This is not noticeable on the posterior pole due to the large amount of coalescent flecks. E, G FAF images of the macular region dating from presentation at proband’s age of 49, 54 and 59 years. The progression of the atrophy is evident. F, H SD-OCT scans passing through the fovea show loss of retinal tissue corresponding to the progression of central atrophy
Given the presence of combinations of severe and mild ABCA4 variants in other cases with late-onset STGD1, we hypothesized that variant c.2653+1G>A results in significant remaining activity of the ABCA4 protein.
c.2653+1G>A results in exon 17 skipping in a midigene splice assay
SpliceAI [43] predicted that c.2653+1G>A weakens the exon 17 splice acceptor site (SAS; delta score 0.63) and splice donor site (SDS; delta score 0.98), and very likely leads to complete skipping of exon 17. To confirm this predicted effect, we included c.2653+1G>A in the previously described BA13 wild-type (WT) midigene [37] that contains ABCA4 exons 16 through 18, and flanking intronic sequences (8258 bp; g.94521462–g.94513205 [GRCh37)]. Although the variant was introduced using a high-fidelity DNA polymerase, the analysis of the entire mutant plasmid sequence identified a single PCR-introduced polymorphism that is unlikely to impact splicing (Additional file 2: Table S3). The rest of the sequence was consistent with previously reported analyses [36]. The expression of constructs was carried out in HEK293T cells that do not express endogenous ABCA4, and the reverse transcription (RT)-PCR of the cDNA confirmed the exon-skipping effect of the variant which was not observed for the WT construct (Fig. 2). As this is a full effect, the protein product is p.Asp864_Gly885del (referred to as ΔAsp864_Gly885 hereafter).

Variant c.2653+1G>A results in complete skipping of exon 17. The ABCA4 genomic sequence flanked by rhodopsin exons 3 and 5. cDNA products were amplified using primers situated in exons 16 and 18, represented with triangles. The mutant construct shows complete exon 17 skipping as shown by Sanger sequencing
ΔAsp864_Gly885 shows partial ABCA4 ATPase activity and localizes to the same cellular structures as wild-type ABCA4
ABCA4 contains a 52 amino acid linker segment (Gly863-His914) encoded by exons 17 and 18 that extends from transmembrane 6 of TMD1 to the start of the NBD1 (Fig. 3A). Within exon 17, residues Gly863-Phe873 could be modeled in the ABCA4 structure in its unbound state (Fig. 3B). In contrast, the remaining linker segment (Leu874-His914) encoded by the remaining part of exon 17 and all of exon 18 was not observed in any of the structures of ABCA4, most likely due to the highly flexible nature of this segment.

Topological and molecular structure of ABCA4. A Topological diagram of ABCA4 showing the various domains of ABCA4 and the location of the peptide segment encoded by exon 17. ECD, exocytoplasmic domain; NBD, nucleotide-binding domain; TMD, transmembrane domain; RD, regulatory domain. Hexagons denote oligosaccharide chains extending from the ECDs. EH, external helix; IH, internal helix. The 22 amino acid-segment encoded by exon 17 is depicted as a red box; B Molecular structure of ABCA4 (PDB: 7LKP). Red sticks show the part of the 22 amino acid-segment encoded by exon 17 that could be modeled (Gly-Asp-Tyr-Gly-Thr-Pro-Leu-Pro-Trp-Tyr-Phe). Right: Enlarged region of ABCA4 containing the segment encoded by exon 17. Dashed line represents the remaining peptide encoded by exons 17 and 18. This segment could not be modeled in any of the ABCA4 structures, presumably due to the flexible nature of this segment of the protein. N-half and C-half are colored blue and orange, respectively
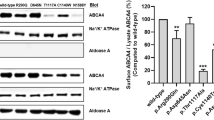
To assess the function of ABCA4 lacking the 22 amino acids encoded by exon 17 variants, ATPase activity of the ΔAsp864_Gly885 protein variant was compared to the activity of wild-type (WT) ABCA4. For these studies, the WT ABCA4 and the ΔAsp864_Gly885 variant were expressed in HEK293T cells and purified by immunoaffinity chromatography. The ΔAsp864-Gly884 variant expressed at similar levels to WT ABCA4 as analyzed on Coomassie blue-stained gels (Additional file 1: Fig. S2) and displayed all-trans retinal (ATR) activated ATPase activity (Fig. 4A). A 1.8 fold increase in ATPase activity in the presence of ATR was observed for WT ABCA4 in agreement with previous studies [39], while the ΔAsp864_Gly884 variant showed a smaller 1.5 fold increase in activity (Fig. 4A). The level of ATR-stimulated activity of the variant was 58.0 ± 4.2% of the activity measured for WT ABCA4. These studies indicate that ABCA4 lacking the peptide segment encoded by exon 17 retains considerable functional activity, a property consistent with a predicted moderate (intermediate) STGD1 phenotype for an individual with a trans null allele based on functional studies [39, 44].

ABCA4 ΔAsp864_Gly885 shows significant activity and displays a subcellular localization in HEK293T cells similar to ABCA4 WT. A ATPase activity of wild-type ABCA4 (WT) and the ΔAsp864_Gly885 variants in the presence and absence of all-trans retinal (ATR) expressed as the percentage activity in the absence of ATR (basal activity). The data is shown as mean ± SEM, n = 3 for each construct and represents 3 independent experiments. B HEK293T cells were transfected with the ABCA4 plasmids, labeled for ABCA4 (red) and calnexin as an ER marker (green) and counterstained with DAPI nuclear stain (blue). The ΔAsp864_Gly885 variant is associated with intracellular vesicles, that resemble vesicles observed for WT ABCA4. A fraction of the truncated protein is retained in the ER
Additionally, the localization of ΔAsp864_Gly885 and of the WT protein was investigated in HEK293T (Fig. 4B) and ARPE-19 cells (Additional file 1: Fig. S2E). Immunocytochemistry analysis revealed that ΔAsp864_Gly885 trafficked to subcellular vesicles, which resembled the localization of the WT protein in both cell types. However, a fraction of the truncated protein co-localized with the endoplasmic reticulum (ER) that was stained with the antibody against calnexin (CNX), as opposed to the WT protein which was not retained in the ER. These results point to a partial misfolding of ΔAsp864_Gly885.
In silico analysis of other in-frame skipped exons in ABCA4
To determine the severity of transcripts lacking in-frame exons other than exon 17 in ABCA4, we performed in silico analyses of canonical SAS and SDS variants in the other 16 exons consisting of a multiple of three base pairs. The effect of these variants on splicing was predicted by SpliceAI and listed in Additional file 3: Table S4. A clear prediction for complete exon skipping was found for all theoretical canonical splice site (CSS) variants located at 11 out of 34 exon–intron junctions in the exons 7, 11, 17, 18, 20, 32, 34 and 49. To assign a severity label to the implicated CSS variants, we consulted published data of previously screened probands for their age of onset and the nature of the ABCA4 variant in trans. We implemented two rules to categorize the CSS variants as severe: 1. the variant in trans was previously classified as mild, and therefore, according to a generally accepted genotype–phenotype model, a second severe allele has to be present in order for STGD1 to occur, or 2. the age of onset was before the age of ten, which generally is found when there are two severe ABCA4 alleles.
According to the analysis in Additional file 4: Table S5, the majority of investigated CSS variants are presumed to lead to ABCA4 transcripts that contain a premature stop codon or frameshift at locations that point to severe effects for these CSS variants. For one reported variant found in homozygosity (c.1554+1G>C) we were unable to find the age of onset, however, the severe cone-rod dystrophy diagnosis allowed to assume the variant’s severe effect. In addition, the amino acid sequences encoded by the 17 in-frame exons were located within the ABCA4 domains to predict their potential role in the structure and function of the protein. Specifically, exons 6, 7, 10, 11, 13, 29, 32 and 34 translate into parts of the ECD1 and ECD2, forming highly interlaced α-helices, glycosylation sites at positions N415, N444 and N504, and a recognition site for N-ret-PE mediated by Arg587 [12, 13]. Other in-frame exons (exons 20, 22, 42, 45 and 47) contribute to α-helical-, β-structures and highly conserved Walker A and B motifs in the NBDs which are characteristic for all proteins that are sustained by ATP-hydrolysis. Exons 15 and 49 encode parts of TMD1 and RD2, respectively (Additional file 5: Table S6). These domains contribute to the correct folding of the protein, as discovered in previous studies [45]. Together with exon 17, exon 18 is predicted to encode the linker that connects TDM1 with NDB1. Unlike other in-frame exons, the amino acids encoded by exons 17 and 18 do not display an available structure, which suggests the presence of a highly flexible region that could potentially endure modifications without causing major disruptions within the structure and function of the protein.
Altogether, this data points to the presumption that in-frame skipping of any of these exons, with the exception of exon 17, is predicted to result in severe or deleterious alleles.
When compared to all ABCA4 CSS variants, we found no statistical differences in frequencies of CSS variants located nearby exons with a multiple of three nucleotides and those located nearby non multiple-of-three nucleotide exons (p = 0.35). The list of all CSS ABCA4 variants can be found in Additional file 6: Table S7.
AON treatment induces the generation of ABCA4 Δexon 17 RNA at significant levels
The LOVD database (https://www.lovd.nl/ABCA4; accessed on 20th January 2023) contains 14 disease-associated variants that reside in exon 17 (Additional file 7: Table S8). Thirteen of these are considered to have a severe effect, as classified previously by Cornelis et al. [17]. Furthermore, most of these variants were classified as “Pathogenic” or “Likely pathogenic” based on ACMG criteria, one is considered “Variant of unknown significance” [c.2617T>A, p.(Phe873Ile)] and one variant could not be categorized [c.2619T>A, p.(Phe873Leu)]. As ABCA4 lacking Asp864_Gly885 shows significant ATPase activity and is partially located in the same cellular structures in HEK293T and ARPE-19 cells, STGD1 patients carrying the above-mentioned variants are likely to benefit from an AON-based treatment that enforces complete skip of exon 17. Thus, an oligo-walk was designed that consisted of 40 AONs that target the entire exon 17 and adjacent introns, and set out to identify the best candidates able to induce the ‘in-frame’ skip. The first screening was carried out in WT WERI-Rb-1 cells with 19 different AONs consisting of 20–22 nt. The molecules were delivered at a 10 µM concentration by gymnosis. AON14 showed the highest efficiency, albeit modest, by leading to 6.4 ± 0.2% of ABCA4 Δexon 17 mRNA and its binding region was defined as the target region for the second AON screening (Additional file 1: Fig. S3). This consisted of a set of 22 AONs of 17–19 nt as previously it was shown that shorter AONs achieved a higher desired effect over longer AONs, likely due to a favored cellular uptake [31]. As shown in Fig. 5A, digital PCR detected 9.0 ± 0.7% of ABCA4 Δexon 17 mRNA isoforms, as opposed to the untreated group where this isoform was present at 0.6 ± 0.1%. As expected, the best AON candidates targeted strong splice enhancer motifs either within the sequence of exon 17 or nearby the downstream splice site, which caused exclusion of the exon (Additional file 1: Fig. S4). Next, these AONs were administered at the same concentration to 190-days-old WT retinal organoids (ROs) and the treatment consisted of a wash-out regimen implemented for 28 days. ABCA4 Δexon 17 mRNA was quantified at significantly higher levels in a range between 32.2 ± 2.1% and 58.8 ± 4.6% post-treatment; the control group treated with a scrambled AON contained no truncated transcript (Fig. 5B). In all groups, the expression of retinal markers (ABCA4, CRX, NRL, NR2E3, and RHO) displayed a consistent pattern, except for OP1NMW that expressed at significantly different levels in ROs treated with AON24 and AON25 when compared to the scrambled AON group (Additional file 1: Fig. S5).

AON-based treatment achieved significant levels of ABCA4 Δexon 17 transcripts. A The percentage of ABCA4 mRNA lacking exon 17 in WT WERI-Rb-1 cells after 48 h long AON treatments, n = 3. B The percentage of ABCA4 mRNA lacking exon 17 in WT retinal organoids is significantly increased upon exposure to the most effective AONs in a 4-week long wash-out treatment, n = 6. The data is shown as mean ± SEM. Asterisks display the significant differences vs. untreated or scrambled AON (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001)
In silico analysis of the off-target effects of the best performing antisense oligonucleotides
The seven AONs used in the treatment of ROs were analyzed for their possible off-target effects in the human genome and transcriptome. As reported in Additional file 8: Table S9, none of the AONs showed full complementarity against the human DNA or mRNA, except for the intended target in ABCA4. Some genomic targets were identified when allowing for one or two mismatches with all AONs, except for AON14 where this was not observed. As the binding location in the genomic targets were either in the intragenic regions or in deep intronic regions ≥ 577 bp distant from the adjacent exon junctions, the AONs are not likely to interfere with the splicing of these genes. However, an exception was identified with AON25, that showed a binding location with 2 mismatches 47 nt upstream the first exon in DNAJC5. In addition, AON27 and AON40 showed a two-mismatch complementarity with the mRNA of RUNDC3A and GALNT5. These genes do not express protein in the retina and therefore are unlikely to be relevant regarding off-target effects of AONs.
Discussion
The main cause underlying STGD1 is reduced or absent phospholipid-retinoid transport exhibited by the ABCA4 transporter, which so far remains without a treatment. Several pre-clinical studies reported promising advances with novel therapies that apply different means to tackle the genetics of the disease, either by reverting a variant’s disease-associated effect on the genomic level with CRISPR-Cas9 or on the level of transcripts by implementing RNA modulating therapies (AONs) [30,31,32,33,34,35, 46]. Despite being too large for introduction in AAV vectors, ABCA4 was successfully delivered and recombined in mouse and pig retinas by combining two AAV vectors and split inteins [47]. All mentioned research is focused on restoring the dysfunction of the diseased protein to its wild-type settings. In this study, we report skipping of the in-frame exon 17 in ABCA4 due to a CSS variant, as observed in a STGD1 proband, that leads to a shortened protein that retained significant levels of ATR-activated basal activity when compared to wild-type ABCA4. This discovery led to the development of potent AONs able to induce skipping of exon 17, which could potentially delay or stop the STGD1 phenotype progression caused by protein-truncating and severe missense variants in ABCA4 exon 17. This novel therapeutic approach, for the first time, implements skipping of a coding region in ABCA4, and widens the arsenal of therapeutic possibilities focused to alleviate the progression of clinical features associated with STGD1.
The ABCA4 gene encompasses 50 exons that is translated into a 2273 amino acid polypeptide. Seventeen of these exons are composed of multiples of three nucleotides. Theoretically, their absence in the mRNA would leave the open reading frame intact, leading to proteins that lack between 11 and 74 amino acids. However, the majority of ‘in-frame’ coding sequences in ABCA4 encode crucial protein domains that are considered essential for the protein’s function. In silico analysis of previously reported CSS variants, predicted to lead to complete skip of numerous in-frame exons, points to their pathogenic nature when taking into consideration the reported allele in trans and onset of age in STGD1 probands (Additional file 1: Table S5). Altogether, the deletion of the structural elements encoded by all other in-frame exons in ABCA4 would, most likely, severely impair the protein’s action, as they play important roles in protein stability, folding, interaction and transport of substrates, and ATP-hydrolysis. Exons 17 and 18 however encode parts of the polypeptide that localize entirely in the linker between TMD1 and NBD1. This region lacks a well-defined structure, and it indicates a highly flexible portion of the protein, which might only be partially essential for the protein’s expression and function. Even though the presence of this seemingly simple element appears to be less substantial when compared to other protein domains, there is little knowledge on its exact function. Of note, a deletion of exon 18 was identified in two individuals [48]. Interestingly, the detected alleles in trans in both cases carried the c.2588G>C p.[Gly863Ala,Gly863del] variant. As c.5603A>T in 2003 was not yet appreciated as a hypomorphic but causal variant, it probably was present in cis with c.2588G>C, and together these two variants form a mild but fully penetrant allele. This finding strongly suggests that skipping of the in-frame exon 18 has a severe effect.
We identified a novel CSS variant downstream exon 17, c.2653+1G>A, in a French STGD1 proband that carried the prior characterized c.735T>G, p.(Tyr245*) variant in trans. Since this case displayed late-onset STGD1, it allowed to hypothesize that c.2653+1G>A leads to translation of ABCA4 with significant remaining functionality. We confirmed the variant’s exon-skipping nature on exon 17 by exploring its expression in midigene-transfected HEK293T cells. The mild nature of the protein lacking the 22 amino acids (ΔAsp864_Gly885) encoded by exon 17 was determined by a functional assay, where the retained ATR-activity was measured at 58% when compared to the WT ABCA4. Furthermore, plasmid-induced overexpression of ABCA4 in HEK293T and ARPE-19 cells showed that ΔAsp864_Gly885 localized in vesicle-like structures previously attributed to the WT ABCA4 [49, 50]. These results suggest that exon 17 is the only in-frame exon that can be omitted from the ABCA4 coding sequence without having a deleterious effect. In addition, the linker structure expressed by this exon most likely has a moderate role in the spectrum of functions exhibited by ABCA4. The significance of this linker for the function of ABCA4 is not known, but it is possible that it serves as a flexible segment that allows ABCA4 to undergo the large conformational change observed upon the binding of ATP to the NBDs during the substrate transport cycle. However, the exact functional role requires further studies.
The data presented above allowed to develop an AON-based treatment able to induce exon 17 skipping in ABCA4. In order to define the patient group that would potentially benefit from this type of therapy, we identified 14 previously reported ABCA4 variants that reside in exon 17. Most of these variants have a deleterious effect on the protein, with the exception of c.2608C>T, p.(Pro870Ser), c.2617T>A, p.(Phe873Ile), c.2617T>C, p.(Phe873Leu) and c.2619T>A, p.(Phe873Leu), for which this is as yet unknown. Nearly all variants in exon 17 are considered “Severe” according to Cornelis et al. [17] and are labelled as “Likely pathogenic” and “Pathogenic” using the ACMG categorization. Thus, STGD1 probands carrying a severe variant in exon 17 would benefit from the AON-mediated exon skip treatment. AONs have emerged as promising therapeutic intervention for treatment of several inherited retinal diseases; two programs continued the investigations in clinical trials exploring the potency of AONs in rescuing the severe phenotypes of Leber congenital amaurosis 10 (LCA10) and USH2A-associated Usher syndrome and retinitis pigmentosa (RP), by applying sepofarsen and ultevursen, respectively. The first clinical trial for sepofarsen showed promising results regarding its safety profile and mechanism of action and identified five out of 11 participants that displayed clinically significant improvements in best corrected visual acuity, full-field stimulus test and mobility course [51]. While sepofarsen removes a pseudoexon, whose insertion is caused by the recurrent c.2991+1655A>G variant, to restore the wild-type splicing in CEP290, ultevursen implements skipping of the in-frame exon 13 in USH2A, which shortens the protein while retaining its residual activity. USH2A-coded usherin contains ten repetitive epidermal growth factor (EGF)-like domains that allow for protein shortening. The AON-mediated ‘in-frame’ exon 13 skipping, which contains frequent and severe USH2A variants, led to a well-tolerated fusion of EGF-like domains 4 and 8; moreover, the results report a properly localized protein with clinically relevant residual activity [26]. As opposed to usherin, ABCA4 lacks repetitive elements whose absence would translate into a protein with residual activity. However, we identified that the lack of Asp864_Gly885 encoded by exon 17 results in a shortened linker between TMD1 and NBD1. Although this approach results in a reduction of ABCA4 activity, the residual activity is still within acceptable levels for use as a therapeutic intervention. Additionally, the AONs described in this study exhibit physicochemical characteristics that are comparable to sepofarsen and ultevursen, which further strengthens their potential for future clinical applications targeting STGD1. However, further investigations into their pharmacological properties are necessary before proceeding with clinical studies.
The results across two AON screenings in a simpler cell model identified candidates that were employed in further screening in a more complex cellular model, 3D human ROs. We observed that the AONs induced higher levels of ABCA4 Δexon 17 transcripts in ROs, as opposed to the effect observed in WERI-Rb-1 cells. This could be justified by the presence of AON-targeted splicing motifs that are most likely acting in a retina-specific manner when contained in a differentiated retinal environment. Previous studies have found that disease-associated isoforms specific to the retinal tissue exhibit differential expression in ROs or photoreceptor precursor cells (PPCs), compared to simpler cell cultures. In fact, the most frequent Leber congenital amaurosis-associated CEP290 variant c.2991 + 1655A > G led to more prominent pseudoexon insertion in patient-derived ROs compared to patient fibroblasts [52]. This trend was also observed for two neighboring ABCA4 intron 30 variants, which created or strengthened exonic splice enhancer motifs, thereby leading to the 345-nt pseudoexon insertion in patient-derived PPCs, but not in patient-derived fibroblasts [53]. Even though WERI-Rb-1 cells represent a common model to investigate AON-mediated suppression of gene expression and exon skipping due to endogenous expression of several IRD-associated genes and retina-like splicing mechanism [26, 54,55,56], ROs are considered to more closely resemble native retinal tissue. Their self-development can be compared to the natural development of the retina [57,58,59], for which reason they became a widely used in vitro model for investigation of disease development and applicable therapeutic compounds [32, 60,61,62,63]. Previously, the ABCA4-targeted AON treatment led to a highly restorative effect on the transcriptional and protein level in both gene-edited and patient-derived ROs. In addition, longer exposition to AONs increased their effect up to twofold when prolonging the treatment from 4 to 8 weeks (31). The extended effect over longer periods can be explained by the inactive portion of oligos entrapped by endosomes and their release over time.
Conclusion
In conclusion, we provide the first evidence of successful exon skipping in ABCA4, resulting in a protein with significant levels of remaining functionality and a correct subcellular localization. Although the coding region of ABCA4 captures multiple in-frame exons whose omission would, in theory, lead to a truncated but functional version of the protein, the high structural organization of ABCA4 presents a challenge for any exon-skipping therapeutic intervention.
While the impact of skipping other ‘in-frame’ exons on the residual activity of ABCA4 is yet unknown, we propose that the remaining ABCA4 activity upon skipping of exon 17 is an exception rather than a rule for any of the other ‘multiple of three nt’ exons. Here, we have shown an innovative AON-mediated therapeutic strategy that can induce skip of the entire exon 17 in ABCA4 and could, potentially, offer therapeutic benefits to STGD1 patients that harbor severe variants in ABCA4 exon 17.
Availability of data and materials
All data generated throughout this study are available within the paper and its supplemental information. Raw data are available upon request from the corresponding author.
Abbreviations
- AAV:
-
Adeno-associated virus
- ABCA4:
-
ATP-binding cassette, subfamily A, member 4
- AON:
-
Antisense oligonucleotide
- cryo-EM:
-
Cryogenic electron microscopy
- CSS:
-
Canonical splice site
- ECD:
-
Extracellular domain
- FA:
-
Fluorescein angiography
- FAF:
-
Fundus autofluorescence
- ffERG:
-
Full field electroretinography
- LCA10:
-
Leber congenital amaurosis type 10
- NBD:
-
Nucleotide-binding domain
- N-ret-PE:
-
N-retinylidene-phosphatidylethanolamine
- RO:
-
Retinal organoid
- RP:
-
Retinitis pigmentosa
- SAS:
-
Splice acceptor site
- SD-OCT:
-
Spectral domain optical coherence tomography
- SDS:
-
Splice donor site
- STGD1:
-
Stargardt disease type 1
- TMD:
-
Transmembrane domain
References
Allikmets R, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–46.
Cremers FPM, et al. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res. 2020;79: 100861.
Cremers FP, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7(3):355–62.
Maugeri A, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000;67(4):960–6.
Runhart EH, et al. Association of sex with frequent and mild ABCA4 alleles in Stargardt Disease. JAMA Ophthalmol. 2020;138(10):1035–42.
Runhart EH, et al. The common ABCA4 Variant p.Asn1868Ile shows nonpenetrance and variable expression of stargardt disease when present in trans with severe variants. Invest Ophthalmol Vis Sci. 2018;59(8):3220–31.
Zernant J, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet. 2017;54(6):404–12.
Hanany M, et al. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci U S A. 2020;117(5):2710–6.
Bungert S, et al. Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J Biol Chem. 2001;276(26):23539–46.
Illing M, et al. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J Biol Chem. 1997;272(15):10303–10.
Liu F, et al. Molecular structures of the eukaryotic retinal importer ABCA4. Elife. 2021;10:e63524.
Scortecci JF, et al. Cryo-EM structures of the ABCA4 importer reveal mechanisms underlying substrate binding and Stargardt disease. Nat Commun. 2021;12(1):5902.
Xie T, et al. Structural basis of substrate recognition and translocation by human ABCA4. Nat Commun. 2021;12(1):3853.
Quazi F, et al. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012;3:925.
Lenis TL, et al. Complement modulation in the retinal pigment epithelium rescues photoreceptor degeneration in a mouse model of Stargardt disease. Proc Natl Acad Sci U S A. 2017;114(15):3987–92.
Cornelis SS, et al. Personalized genetic counseling for Stargardt disease: Offspring risk estimates based on variant severity. Am J Hum Genet. 2022;109(3):498–507.
Cornelis SS, et al. In silico functional meta-analysis of 5962 ABCA4 variants in 3928 retinal dystrophy cases. Hum Mutat. 2017;38(4):400–8.
Bayever E, et al. Systemic administration of a phosphorothioate oligonucleotide with a sequence complementary to p53 for acute myelogenous leukemia and myelodysplastic syndrome: initial results of a phase I trial. Antisense Res Dev. 1993;3:383–90.
Cunningham CC, et al. A phase I trial of c-Raf kinase antisense oligonucleotide ISIS 5132 administered as a continuous intravenous infusion in patients with advanced cancer. Clin Cancer Res. 2000;5:1626–31.
Henry SP, et al. Evaluation of the toxicity of ISIS 2302, a phosphorothioate oligonucleotide, in a 4-week study in CD-1 mice. Antisense Nucleic Acid Drug Dev. 1997;7(5):473–81.
Webb A, et al. BCL-2 antisense therapy in patients with non-Hodgkin lymphoma. Lancet. 1997;349(9059):1137–41.
Yuen AR, et al. Phase I study of an antisense oligonucleotide to protein kinase C-α (ISIS 3521/CGP 64128A) in patients with cancer. Clin Cancer Res. 1999;5(11):3357–63.
Mulamba GB, et al. Human cytomegalovirus mutant with sequence-dependent resistance to the phosphorothioate oligonucleotide fomivirsen (ISIS 2922). Antimicrob Agents Chemother. 1998;42(4):971–3.
Frank DE, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94(21):e2270–82.
Mendell JR, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–47.
Dulla K, et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol Ther. 2021;29(8):2441–55.
ProQR Therapeutics. Study to evaluate the efficacy safety and tolerability of QR-421a in subjects with RP due to mutations in exon 13 of the USH2A gene with early to moderate vision loss (Celeste). https://clinicaltrials.gov/ct2/show/NCT05176717. Accessed 9 Jan 2023.
ProQR Therapeutics. Study to Evaluate the Efficacy Safety and Tolerability of Ultevursen in Subjects With RP Due to Mutations in Exon 13 of the USH2A Gene (Sirius). https://clinicaltrials.gov/ct2/show/NCT05158296. Accessed 9 Jan 2023.
Bauwens M, et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet Med. 2019;21(8):1761–71.
Garanto A, et al. Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c4539+2001G>A in Stargardt disease. Genes. 2019;10(6):452.
Kaltak M, et al. Antisense oligonucleotide therapy corrects splicing in the common Stargardt disease type 1-causing variant ABCA4 c.5461–10T>C. Mol Ther Nucleic Acids. 2023;31:674–88.
Khan M, et al. Detailed phenotyping and therapeutic strategies for intronic ABCA4 variants in Stargardt disease. Mol Ther Nucleic Acids. 2020;21:412–27.
Sangermano R, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019;21(8):1751–60.
Tomkiewicz TZ, et al. Correction of the splicing defect caused by a recurrent variant in ABCA4 (c.769–784C>T) that underlies Stargardt disease. Cells. 2022;11(24):3947.
Tomkiewicz TZ, et al. Antisense oligonucleotide-based rescue of aberrant splicing defects caused by 15 pathogenic variants in ABCA4. Int J Mol Sci. 2021;22(9):4621.
Sangermano R, et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018;28(1):100–10.
Gamundi MJ, et al. Transcriptional expression of cis-acting and trans-acting splicing mutations cause autosomal dominant retinitis pigmentosa. Hum Mutat. 2008;29(6):869–78.
Sangermano R, et al. Photoreceptor progenitor mRNA analysis reveals exon skipping resulting from the ABCA4 c.5461–10T–>C mutation in Stargardt disease. Ophthalmology. 2016;123(6):1375–85.
Curtis SB, et al. Functional analysis and classification of homozygous and hypomorphic ABCA4 variants associated with Stargardt macular degeneration. Hum Mutat. 2020;41(11):1944–56.
Ahn J, et al. Functional interaction between the two halves of the photoreceptor-specific ATP binding cassette protein ABCR (ABCA4). Evidence for a non-exchangeable ADP in the first nucleotide binding domain. J Biol Chem. 2003;278(41):39600–8.
Hallam D, et al. Human-induced pluripotent stem cells generate light responsive retinal organoids with variable and nutrient-dependent efficiency. Stem Cells. 2018;36(10):1535–51.
Stenirri S, et al. Denaturing HPLC profiling of the ABCA4 gene for reliable detection of allelic variations. Clin Chem. 2004;50(8):1336–43.
Jaganathan K, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535-48 e24.
Molday RS, et al. Structure and function of ABCA4 and its role in the visual cycle and Stargardt macular degeneration. Prog Retin Eye Res. 2021;89:101036.
Zhong M, et al. Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J Biol Chem. 2009;284(6):3640–9.
De Angeli P, et al. Effective splicing restoration of a deep-intronic ABCA4 variant in cone photoreceptor precursor cells by CRISPR/SpCas9 approaches. Mol Ther Nucleic Acids. 2022;29:511–24.
Tornabene P, Trapani I, Minopoli R, Centrulo M, Lupo M, de Simone S, Tiberi P, Dell’Aquila F, Marrocco E, Iodice C, Iuliano A. Intein-mediated protein trans-splicing expands adeno-associated virus transfer capacity in the retina. Sci Transl Med. 2019;11(492):p.eaav4523.
Yatsenko AN, et al. An ABCA4 genomic deletion in patients with Stargardt disease. Hum Mutat. 2003;21(6):636–44.
Garces F, et al. Correlating the expression and functional activity of abca4 disease variants with the phenotype of patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2018;59(6):2305–15.
Garces FA, et al. Functional characterization of ABCA4 missense variants linked to Stargardt macular degeneration. Int J Mol Sci. 2020;22(1):185.
Russell SR, et al. Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: a phase 1b/2 trial. Nat Med. 2022;28(5):1014–21.
Dulla K, et al. Splice-Modulating Oligonucleotide QR-110 Restores CEP290 mRNA and function in human c.2991+1655A>G LCA10 Models. Mol Ther Nucleic Acids. 2018;12:730–40.
Albert S, et al. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying stargardt disease. Am J Hum Genet. 2018;102(4):517–27.
Boatright JH, et al. Endogenous CRX expression and IRBP promoter activity in retinoblastoma cells. Brain Res. 2001;916:136–42.
Grayson C, et al. Retinoschisin, the X-linked retinoschisis protein, is a secreted photoreceptor protein, and is expressed and released by Weri–Rb1 cells. Hum Mol Genet. 2000;9(12):1873–9.
Suarez-Herrera N, et al. Development and use of cellular systems to assess and correct splicing defects. In: Arechavala-Gomeza V, Garanto A, editors., et al., Antisense RNA design, delivery, and analysis. Humana; 2022. p. 145–65.
Eiraku M, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472(7341):51–6.
Nakano T, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10(6):771–85.
Zhong X, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun. 2014;5:4047.
Buskin A, et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat Commun. 2018;9(1):4234.
Deng WL, et al. Gene correction reverses ciliopathy and photoreceptor loss in ipsc-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Reports. 2018;10(4):1267–81.
Lukovic D, et al. Retinal organoids derived from hiPSCs of an AIPL1-LCA patient maintain cytoarchitecture despite reduced levels of mutant AIPL1. Sci Rep. 2020;10(1):5426.
Parfitt DA, et al. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell. 2016;18(6):769–81.
Acknowledgments
We are grateful to the patient for participating in this study. We thank Stéphanie S. Cornelis (RUMC) for providing insights in the ACMG classification of severe exon 17 variants. We thank Hee Lam Chan (ProQR Therapeutics) for characterizing and supplying retinal organoids. We thank Marko Potman (ProQR Therapeutics) for providing ARPE-19 cells. Figures and the graphical abstract were designed using bioRENDER.
Funding
This study was financed through European Union's Horizon 2020 research and innovation programme Marie Sklodowska-Curie Innovative Training Networks (ITN) under grant No. 813490 (StarT) and supported by a Canadian Institutes of Health Research (CIHR) grant (PJT 175118).
Author information
Authors and Affiliations
Contributions
MK, JS, and FPMC conceptualized the study. JS, RSM, and FPMC supervised the project. MK, RBG, LLM, CMD, JS, RSM, and FPMC set up the experiment design. MK, RBG, LLM, CMD, EES, RSM, and FPMC performed data analysis. MK, RBG, RSM, and FPMC wrote the original draft. MK, LLM, CMD, JS, RSM, and FPMC contributed to the revisions. GP, RSM, and FPMC assisted in funding acquisition. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
For collecting human samples, written informed consent was obtained before participation. These studies were conducted in accordance with French Law No. 2011-814 of July 7, 2011, Decree 2013-527 of June 20, 2013 and Order of May 27, 2013.
Consent for publication
All authors have read and approved the content of the manuscript and agree to submit for publication in the journal.
Competing interests
M.K., G.P., and J.S. were employed by ProQR Therapeutics during this project. M.K., and J.S. are inventors on the international patent application (WO 2022/090256) that has been filed by ProQR Therapeutics describing methods and means related to exon 17 skip of ABCA4 pre-mRNA by antisense oligonucleotides for treatment of Stargardt disease type 1. The rest of the authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Proband’s baseline full field electroretinography (ffERG) results (ISCEV protocol; MetroVision, Perenchies, France). A Moderately subnormal scotopic rod (dark adapted) responses for both left (OS, oculus sinister) and right eyes (OD, oculus dexter). B Moderately subnormal mixed response (dark adapted), of which the right eye shows a more diminished response. C Subnormal oscillatory responses (dark adapted) for both eyes (OP, oscillatory potential). D Very diminished cone responses (light adapted) in both eyes. E Subnormal flicker response (light adapted) for both eyes. Figure S2. Expression of ABCA4 constructs in HEK293T (A, C) and ARPE-19 (B, D, E) cells. A Coomassie blue stained gel of affinity purified wild-type (WT) and ΔAsp864_Gly885 proteins. B Immunoblot of wild-type (WT) and ΔAsp864_Gly885 proteins from ARPE-19 protein lysates. Vinculin (VCL) was used as loading control. C, D Quantitation showed that ABCA4 ΔAsp864_Gly885 expressed at levels similar to those measured for the WT ABCA4 in both HEK293T and ARPE-19 cells, respectively. Data are shown as mean±SEM. E ARPE-19 cells were transfected with the ABCA4 plasmids, and the localization of ABCA4 was detected using the ABCA4 3F4 clone (yellow). The ER, detected with anti-calnexin (CNX), is shown in magenta. ΔAsp864_Gly885 is associated with intracellular vesicles, that resembles the localization of the WT. Part of the truncated protein is retained in the ER, which was not identified with WT protein. Scale 20 μM. Figure S3. Percentages of ABCA4 Δexon 17 transcripts upon treatment with AONs in the first screening. The AONs were administered to WERI-Rb-1 cells gymnotically and the treatment lasted 48 h. Data are shown as mean±SEM, **p≤0.01, ****p≤0.0001 vs. untreated. Figure S4. Binding location of antisense oligonucleotides in first and second screening. Capital letters display ABCA4 exon 17. Colored horizontal lines represent the binding sites for RNA-splicing proteins (adapted from Alamut Visual Plus). The AONs in red represent the best candidates that were used in treatment of retinal organoids. Figure S5. The expression of retinal markers in retinal organoids (ROs) treated with AONs. All markers show similar expressions between all groups of ROs, except for OPN1MW that showed significant differences in ROs treated with AON24 and AON25 vs. scrambled group. Data are shown as mean±SEM. Asterisks indicated the significant differences in expression vs. scrambled, **p≤0.01, ***p≤0.001. Table S1. List of antisense oligonucleotides. Table S2. List of primers and probes used in dPCR assays.
Additional file 2:
Table S3. Polymorphisms identified in the BA_13 plasmid upon mutagenesis.
Additional file 3:
Table S4. SpliceAI predictions for the effect on splicing of canonical splice site variants.
Additional file 4:
Table S5. The canonical splice site variants predicted to lead to complete exon skip with reported variants in trans.
Additional file 5:
Table S6. The in-frame exon localization and role within the ABCA4 protein.
Additional file 6:
Table S7. Data analysis for pathogenicity and severity allocation.
Additional file 7:
Table S8. ABCA4 exon 17 variants.
Additional file 8:
Table S9. In silico analysis of possible intergenic, intronic and coding off-targets with maximum of two mismatches in AONs used in retinal organoid treatment.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kaltak, M., Blanco-Garavito, R., Molday, L.L. et al. Stargardt disease-associated in-frame ABCA4 exon 17 skipping results in significant ABCA4 function. J Transl Med 21, 546 (2023). https://doi.org/10.1186/s12967-023-04406-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04406-x