
Abstract
Immunotherapy for cancer is making impressive strides at improving survival of a subset of cancer patients. To increase the breadth of patients that benefit from immunotherapy, new strategies that combat the immunosuppressive microenvironment of tumors are needed. Phosphatidylserine (PS) signaling is exploited by tumors to enhance tumor immune evasion and thus strategies to inhibit PS-mediated immune suppression have potential to increase the efficacy of immunotherapy. PS is a membrane lipid that flips to the outer surface of the cell membrane during apoptosis and/or cell stress. Externalized PS can drive efferocytosis or engage PS receptors (PSRs) to promote local immune suppression. In the tumor microenvironment (TME) PS-mediated immune suppression is often termed apoptotic mimicry. Monoclonal antibodies (mAbs) targeting PS or PSRs have been developed and are in preclinical and clinical testing. The TIM (T-cell/transmembrane, immunoglobulin, and mucin) and TAM (Tyro3, AXL, and MerTK) family of receptors are PSRs that have been shown to drive PS-mediated immune suppression in tumors. This review will highlight the development of mAbs targeting PS, TIM-3 and the TAM receptors.
Video Abstract
Similar content being viewed by others

Background
Michele Peyrone in 1845 described a molecule that had anti-cancer activity called “Peyrone salt,” Alfred Werner in 1893 deduced the structure of the salt, and Barnett Rosenberg in 1965 discovered the biological effects of this salt, a substance that the field of oncology now knows as cisplatin [1, 2]. Since 1965, life-changing advancements in chemotherapy design and utilization have been made but hurdles for the systemic treatment of cancer remain. The realization that the immune system can be harnessed to fight a patients’ own disease has provided a new arsenal of strategies for cancer therapy [3,4,5,6,7,8,9,10,11]. Immunotherapy is now first line therapy for some cancers [12,13,14,15] and the immunotherapy options have grown substantially, to include vaccines, immune checkpoint blockade, immune agonists and chimeric antigen receptor (CAR) T-cell therapy [16,17,18,19]. To expand the impact of immunotherapy, signaling pathways that drive tumor evasion of immune surveillance are under robust investigation. Phosphatidylserine (PS), an anionic phospholipid present in all mammalian cells has been studied for the past two decades as a critical immunosuppressive feature that tumors use to mask their presence from the immune system. Research has shown targeting PS or PS-receptors (PSR) with monoclonal antibodies (mAb) can alter PS-mediated immunosuppression and facilitate the induction of an innate and adaptive anti-tumor immune response. This review will cover the current literature of targeting PS and PSRs by monoclonal antibodies for the treatment of cancer.
Phosphatidylserine
Lipid bilayers envelop eukaryotic cells and organelles to subdivide the cell into distinct working compartments. Phospholipid bilayers account for almost three-quarters of mammalian cell content. The major phospholipids in the cell include phosphatidylcholine (PC) and phosphatidylethanolamine (PE), which make up 45–50% and 30–40% of the phospholipids in cell, respectively. Other phospholipids, which are less abundant but integral to membrane function and homeostasis are phosphatidylinositol (PI), PS, and phosphatidic acid (PA) [20, 21]. While PS is a minor constituent in eukaryotic cells, PS-induced processes are highly conserved and have significant physiological functions.
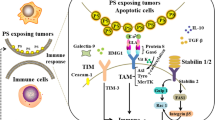
PS is asymmetrically distributed to the inner leaflet of the plasma membrane in a highly conserved ATP-dependent process [22, 23]. PS is redistributed or flipped to the outer leaflet of the plasma membrane during or as result of certain cellular contexts or processes, the most well-described of which is apoptosis [24]. PS redistribution during apoptosis is facilitated by floppases and scamblases [24, 25]. TMEM16F is a Ca2+-dependent membrane associated phospholipid scramblase that can translocate PS to the outer leaflet of the plasma membrane [24]. However, TMEM16F is not required for exposure of PS in apoptotic cells. XKR8 is a caspase 3/7-activated phospholipid scramblase that appears to be responsible for PS exposure as a consequence of apoptosis [24]. Other scramblases, members of the TMEM16 and XKR families also exist and may function in a tissue selective manner and/or function as alternative scramblases that translocate PS [24]. Apoptosis induction and membrane phospholipid asymmetry collapse can be caused by perturbations in ion (Ca2+, K+, Na+) channels, the production of reactive oxygen species (ROS) via cell stress or mitochondrial-initiated apoptosis and caspase activation via DNA damage, radiation damage, and metal toxicity [26]. After PS is redistributed to the cell surface it can function as an “eat me” signal that initiates efferocytosis [27]. Aside from externalization on apoptotic bodies, PS has also been reported to be externalized on other cell types such as immune cells and cancer cells. For example, PS is found on myeloid-derived suppressor cells (MDSCs), monocytes, macrophages, active B cells, dendritic cells (DCs) activated mast cells and T cells [28,29,30,31,32,33,34]. In the tumor microenvironment (TME), exposed PS can also be found on tumor cells, secreted microvesicles and tumor endothelial cells [35]. PS-mediated efferocytosis initiates a highly conserved process that prevents local and systemic immune activation via signaling by PSRs. Importantly, PSR activation on immune cells creates an immunosuppressive milieu that tumor cells use as immune-camouflage [28]. Immune cells including MDSCs, CD4+ and CD8+ T cells, DCs, macrophages, B cells, and natural killer cells (NKs) express PSRs [36, 37].
PSRs are separated into two distinct families: those that bind PS directly and those that bind PS via a bridging protein (see Table 1). PSRs that are direct PS binders are exemplified by the T cell/transmembrane, immunoglobulin, and mucin (TIM) family of receptors, which are well characterized for their immune regulatory activity driven by PS binding [38, 39]. PSRs that are indirect PS binders are exemplified by the Tyro3, AXL, and MerTK (TAM) receptor tyrosine kinase (RTK) family that use gamma carboxylated growth arrest-specific 6 (Gas6) and Protein S (ProS) as the bridging molecule linking the receptor to PS [40]. TAM receptors have also been characterized for immune regulatory activity after PS-induced activation [15, 36, 40]. Given that PS-mediated signaling can induce local immune suppression and that tumors exploit this evolutionarily conserved pathways to evade immune detection, it is reasonable to suggest that interfering with PSR activity could augment anti-cancer immune therapy. Multiple strategies to interfere with PSR activity have been developed including monoclonal antibodies (mAbs) that target PS [41, 42].
Background and current developments with mAb immunotherapy targeting
TIM-3
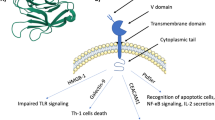
In humans there are three genes that make up the TIM family: TIM-1, − 3, and − 4 [43]. TIM genes encode type 1 membrane spanning proteins and TIM receptors consist of four well-defined regions: the variable immunoglobulin domain (IgV), mucin domain, transmembrane region, and intracellular stem [44]. All 3 TIM receptors have been implicated as PSRs [45, 46]; however, inhibitory TIM-3 mAbs are further advanced and will be discussed here. It should be noted that no current TIM-1 or TIM-4 mAb clinical trials are ongoing although antibody-drug conjugates (ADCs) targeting these receptors are being developed [47]. TIM-3 is expressed in multiple types of cancers including sarcoma, cervical and gastric cancer, myeloma, melanoma, and lung cancer [43, 48,49,50,51] and expression of TIM-3 correlates with worse outcome [43, 44, 46]. TIM-3 is also expressed on different immune cell types. For example, TIM-3 has been reported on DC populations, which suggests that antigen presentation and phagocytosis can be affected by this PSR [52]. TIM-3 expression is also found on CD8+ T cells, regulatory T cells (Tregs), and NK cells [50]. Furthermore, M2-like macrophages show higher levels of TIM-3 expression than M1-like macrophages [44, 53]. Consistent with human expression data, TIM-3 expression on peripheral blood monocytes and tumor-associated macrophages has been shown to correlate with disease progression in a murine model of hepatocellular carcinoma [44, 54]. Interestingly, PS is the only nonprotein known to bind to the family of TIM receptors. It should be noted aside from PS, TIM-3 has been identified to interact with several other proteins implicated in immune regulation, including galectin-9 (gal-9), carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1), and high-mobility group protein box 1 (HMGB-1) [14, 55,56,57,58,59]. PS binding directly to TIM-3 has been confirmed and it has been shown to induce efferocytosis in phagocytic cells [60] although the affinity of TIM-3 for PS is weaker than TIM-1 and 4 [61] [62]. It has been proposed that PS and TIM-3 interactions promote immune cell exhaustion since PS is involved in immune cell tolerance. Silva et al. working on reversing NK cell exhaustion hypothesized that since PS is on the surface on apoptotic bodies, it might stimulate NK cell exhaustion after effector-induced tumor cell death [63]. In addition, TIM-3+ APCs phagocytize apoptotic bodies but T-cells that express TIM-3 form conjugates that are not capable of phagocytosis. However, Freeman et al. proposes that cross-linking conjugates on T-cells by apoptotic bodies may provide an immunostimulatory signal to T-cells [45]. This effect would be induced because of the binding of TIM-3 on Th1 or Th17 cells via galectin-9 [64, 65].
Immune cells that express TIM-3 promote immune tolerance to tumors and thus therapeutic mAbs that target TIM-3 have been developed and tested preclinically and clinically. Studies in multiple animal models have shown that antibody-mediated Tim-3 inhibition enhances the activity of immune checkpoint blockade [66,67,68] although detailed analysis of the tumor immune landscape is still incomplete after Tim-3 inhibition. High levels of TIM-3 correlate with exhausted CD8+ T cells in melanoma patients and anti–TIM-3 mAb treatment reversed this phenotype [69]. Non–small-cell lung cancer (NSCLC) patients were found to have high expression of TIM-3 on CD4+ and CD8+ T cells [69]. TIM-3 has also been found on tumor-infiltrating lymphocytes (TILs) in head and neck cancer, renal cell carcinoma, gastric cancer, non-Hodgkin’s lymphoma, cervical cancer, prostate cancer, colorectal cancer, and hepatocellular cancer [70]. Furthermore, TIM-3 expression is now recognized as a marker of T cell exhaustion. This is illustrated by a recent study, where TIM-3+ TILs co-expressed programmed cell death protein 1 (PD-1) and lacked interleukin-12 (IL-12), tumor necrosis factor (TNF), and interferon gamma (IFNy) expression [68, 71]. This has ignited speculation that combining anti-TIM-3 with anti–PD-1 therapy might be a viable option to overcome T-cell exhaustion in patients and promote responses to immune checkpoint blockade. Furthermore, TIM-3 inhibition has been implicated as a possible strategy for priming response to other therapies such as Toll-like receptors (TLR) agonists to promote an active anti-tumor immune response. For example, blocking TIM-3 followed by TLR agonist treatment resulted in the expression of IL-12, interleukin-10 (IL-10), and interleukin-6 (IL-6) in hepatitis C monocytes, and this strategy may be applicable to cancer [72].
Antibodies against TIM-3 are being investigated in multiple clinical trials (see Table 2). NCT03680508 is a phase II trial, testing anti–TIM-3 mAb TSR-022 in combination with anti–PD-1 mAb TSR-042 in patients with hepatocellular carcinoma [73]. Early data suggests that blocking TIM-3 enhances cytotoxic T-cell–mediated tumor lysis [74, 75]. NCT02608268 is studying the effect of anti-TIM-3 in advanced malignancies. This phase I/II trial is evaluating anti–TIM-3 mAb as a single agent and in combination with PDR001 (anti–PD-1 antibody).
TAM receptors
TAM receptors contribute to cancer development, growth and metastasis. The two most characterized TAM ligands are vitamin K-dependent proteins, Gas6 and ProS [76]. Gas6 and ProS bind PS via gamma carboxylation motif and are produced by multiple cell types, including tumor cells, immune cells and fibroblasts in the TME [77, 78]. TAM receptors expressed by phagocytic cells participate in efferocytosis and can induce a tolerogenic immune cell phenotype [79,80,81], thereby promoting tumor immune evasion. For example, TAM receptors have been found on macrophages, DCs, NK cells, T cells, and can indirectly affect T-cell functions in the TME [81]. Axl and MerTK are expressed in bone marrow-derived DCs and Gas6 has been shown to mediate reduced TLR response as measured by production of IL-6, tumor necrosis factor alpha (TNFα), and type I interferon after TLR agonist stimulation [81, 82]. In addition, Axl activation on macrophages and DC can result in the upregulation of negative TLR and cytokine regulators, suppressor of cytokine signaling-1 (SOCS1) and suppressor of cytokine signaling-3 (SOCS3), which further dampen immune activation [83]. Mouse models have shown that a lack of expression of TAM receptors or inhibition of TAM signaling can increase immune-mediated rejection of tumor cells [84, 85]. Additionally, TAM receptors prevent the induction of immune responses by preventing the activation of antigen-presenting cells (APCs) via PS binding with Gas6 or ProS [86]. TAM receptors, Axl and MerTK, are also expressed by tumor cells in many tumor types [81]. Activation of Axl/MerTK on tumor cells results in induction and maintenance of a mesenchymal-like tumor cell phenotype.
As a result, TAM receptors can drive epithelial plasticity or epithelial to mesenchymal transition (EMT) [40]. EMT is linked to tumor cell survival, therapy resistance, metastasis and immune suppression in multiple tumor types [87, 88]. Multiple strategies to inhibit TAM receptors have been developed. These include neutralizing mAbs, ADCs and small molecule inhibitors. Recent reviews on the validation of Axl and MerTK as therapeutic targets are available (78, Parinot, 2016 #145). Here we will provide an overview of mAbs targeting TAM receptors and how these agents impact the tumor microenvironment.
Pre-clinical studies with mAb targeting the TAM receptors have contributed to our understanding of the function of TAM receptors in cancer. Antibodies discussed in this section are shown in Table 3. Demarest et al. [89] published a robust study on a series mAbs specific for Tyro3 in melanoma cell lines. They identified mAbs that show moderate to high affinity to the extracellular domain of Tyro3 and a range of activity in blocking Gas6 binding to the receptor and inhibition of ligand-induced Tyro3 signaling. Chien et al. [90] engineered a human anti-Tyro3 mAb, Tyro3-hIgG, and reported that the mAb inhibited cell migration and invasion in human colon cancer cells and NIH3T3 fibroblasts. They also provided evidence that inhibition of Tyro3 can reverse EMT and enhance sensitivity of cancer cells to chemotherapy. These findings along with multiple other studies [91,92,93,94,95,96] have highlighted the contribution of Tyro3 to the tumor microenvironment. To our knowledge, Tyro3 specific mAbs have not advanced to clinical testing to date.
In contrast to Tyro3, numerous groups have developed mAbs specific for Axl. Multiple preclinical studies with the Axl mAb DAXL-88 have shown that it can inhibit tumor cell migration and invasion in vitro [97]. In addition, DAXL-88, which binds mouse and human Axl has shown impressive anti-tumor efficacy in mice bearing MDA-MB-231 xenografts [97]. BA3011 is another Axl targeting mAb that selectively binds to human Axl [98]. BA3011 showed efficacy in lung, prostate and pancreatic cancer xenograft models [98] and has been developed as an ADC, CAB-AXL-ADC with a proprietary protein as the drug. CAB-AXL-ADC has entered clinical testing (trial identifier NCT0342527). Other therapeutic anti-Axl mAbs that have shown efficacy in preclinical models of cancer include YW327.6S2 (YW) and 20G7-D9. YW is a phage-derived mAb that showed anti-tumor efficacy in preclinical models of NSCLC and breast cancer models [99]. YW recognizes mouse and human Axl [99], inhibits the binding of Gas6 to Axl in a dose-dependent–mediated manner and downregulates Axl receptor expression. In xenograft studies, YW reduced vascular density and inhibited inflammatory cytokine expression from tumor-associated macrophages [99]. YW also enhanced the efficacy of EGFR inhibition with erlotinib in NSCLC xenografts [100] and reduced metastasis [99]. Clinical studies with YW are likely and could include combination with anti-vascular endothelial growth factor (VEGF) strategies. 20G7-D9, has also been evaluated in multiple breast cancer models, including xenograft and patient-derived xenografts [101]. 20G7-D9 inhibited tumor growth and bone metastasis lesions in a tumor cell Axl-dependent manner, highlighting the importance of tumor cell Axl expression to tumor progression and the efficacy of Axl targeted agents [101]. In addition, 20G7-D9 induced Axl degradation and inhibited Gas6-dependent cell signaling, cell migration and EMT [101]. 20G7-D9 is being developed as a therapeutic mAb and an ADC. Axl mAbs are currently being evaluated in preclinical studies in combination with immune therapy in multiple indications.
Antibodies specific for MerTK have also been developed and tested in preclinical cancer models. RGX-019 is a MerTK targeting mAb that prevents Gas6 induced phosphorylation of AKT resulting in inhibition of melanoma cell growth and colony formation [102]. In addition, the same study showed RGX-019 prevented MDA-MB-231 breast tumor growth in vivo. Cummings et al. [103] reported on another MerTK targeting mAb, Mer590 that reduced MerTK levels in NSCLC cell lines in vitro. Mer590 inhibited STAT6, AKT and ERK1/2 activation and resulted in MerTK down-regulation, resulting in increased apoptosis and decreased colony formation.
At the time of writing this review there are no active clinical trials involving mAbs targeting TAM receptors; however, it is anticipated that multiple TAM mAbs will enter clinical testing soon. Preclinical studies with small molecular weight inhibitors of MerTK and Axl have been shown to alter the tumor immune landscape to favor anti-tumor immune activity [77, 104, 105], thus it is anticipated that antibody-mediated inhibition of TAM receptors will also alter the tumor immune landscape. TAM receptors have a clear function in immunosuppressive signaling in cancer and it is likely that mAbs targeting TAMs will be evaluated in the context of immune checkpoint blockade in cancer patients.
Phosphatidylserine
Antibodies that target PS were developed by Philip Thorpe’s laboratory to specifically home to tumor vasculature (reviewed in Belzile [27]). The realization that PS is externalized on tumor endothelial cells but not on endothelial cells in normal tissues was the result of studies on the efficacy of a coagulation-inducing vascular targeting agent (VTA) specific for vascular cell adhesion molecule 1 (VCAM1) in tumor-bearing mice [106]. Ran et al. [106] showed that a VCAM1-targeted VTA localized to tumor blood vessels and vessels in some normal organs, including cardiac blood vessels; yet coagulation was only induced in the tumor. They went on to demonstrate that the selective efficacy was due to exposure of PS on the luminal surface of tumor endothelial cells, which supported initiation of the coagulation cascade. This led to the development of a series of mAbs that target PS. Unlike other PS binding agents, including annexin V, the mAbs bind PS in a calcium-independent manner [106,107,108]. Robust in vivo localization studies in mice revealed that PS-targeting mAbs and annexin V specifically localize to tumor vasculature but were not present normal organs evaluated [27, 106,107,108]. These observations suggested that targeting anionic lipids, such as PS, was viable and potentially useful as an anti-cancer strategy.
The majority of PS-targeting mAbs developed by the Thorpe laboratory bind PS via a serum cofactor, β2 glycoprotein 1 (β2GP1) (see Table 4). β2GP1, a known PS interacting protein [109, 110], is a 5-domain protein found in abundance in sera (~ 200 μg/mL). In its native state, β2GP1 is in a circular protein conformation [111]. Studies indicate β2GP1 adopts an open “J-shape” structure in the presence of select antibodies and other activating proteins/lipids [110]. PS-targeting mAbs, including 3G4 and its derivatives, bind and dimerize β2GP1 such that domain 5 of each β2GP1 in the complex binds with high affinity to PS on the plasma membrane [27]. Figure 1 presents a schematic depicting the interaction of PS-targeting mAbs with β2GP1 and PS. The PS-targeting mAbs while initially developed to selectively bind to tumor vasculature were found to have anti-tumor efficacy in preclinical tumor models [112]. In fact, the mAbs have been shown to enhance the efficacy of standard chemotherapy [58, 113, 114] and radiation therapy [115, 116] in multiple mouse cancer models. β2GP1 is the primary antigen associated with anti-phospholipid syndrome, an autoimmune disorder characterized by the production of anti-phospholipid antibodies that enhance thrombosis and complications during pregnancy and is associated with systemic lupus erythematosus. Mineo et al. found that one of the Thorpe PS-targeting mAbs (1 N11) prevents the pathogenesis induced by anti-phospholipid antibodies in preclinical models [117]. These data suggest that not all antibodies that bind β2GP1 are the same and also highlight that 1 N11 or other therapeutic anti-PS targeting mAbs might have utility for the treatment of APS.

Diagram depicting PS-targeting mAb and β2GP1 binding to PS on a cell surface. Proposed mechanism of PS-targeting mAb binding to exposed PS in a β2GP1-dependent manner. Not drawn to scale
Investigation of the anti-cancer mechanism(s) of action of PS-targeting mAbs showed that 40% of blood vessels were bound by the mAb in orthotopic human breast xenografts [112]. Additionally, the mAb appeared to induce antibody dependent cellular cytotoxicity (ADCC) directed towards endothelial cells and this effect was magnified in the presence of chemotherapy [113]. These results suggest that chemotherapy induces increased PS externalization and that PS-targeting mAbs alter immune cell phenotype as macrophages in the TME typically are not capable of performing ADCC. Evidence supporting that PS-targeting can alter immune cell phenotype includes the observation that tumor vasculature was reduced after treating with a PS-targeting mAb + docetaxel and this corresponded to a 4 and 14-fold increase in macrophage infiltration into tumors treated with a PS-targeting mAb alone or in combination with docetaxel, respectively [113]. PS-targeting also enhanced the efficacy of PRIMA-1 (APR-246), a therapeutic agent that reactivates mutant p53 [118, 119]. In addition, PS-targeting showed similar anti-tumor efficacy when combined with an onco-adenovirus, Delta-24-RGD, that replicates in tumors and promotes high PS exposure after viral infection [120]. Supporting these observations, additional studies provided evidence that PS-targeting alters myeloid cell phenotype in human tumor xenografts. Yin et al. [121] found that PS-targeting mAbs dramatically shifted the phenotype of macrophages from an M2-like to a M1-like phenotype and that the mAb induced the differentiation of MDSCs to M1-like macrophages and mature DCs and reduced the expansion of immunosuppressive cell types, including MDSCs and Tregs in the TME [121]. Additionally, the authors demonstrated through electron microscopy that the PS-targeting mAbs interact with immune cells through extracellular vesicles and also provided evidence that this immune reprogramming is dependent on the Fc portion of the PS-targeting mAb suggesting that the change in immune cell phenotype is dependent upon a) blocking PS-PSR interaction and b) Fc receptor engagement on the immune cell. One of the key consequences of PS-targeting mAb activity is DC maturation, which can presumably impact induction of an adaptive immune response.
The first evidence that PS-targeting mAbs could facilitate an adaptive immune response was shown by He et al [115]. The authors found that radiation in combination with a PS-targeting mAb induced long-term survival in rats bearing orthotopic syngenic F98 glioma cells. Additionally, splenocytes from long-term survivors showed cytotoxic activity against F98 tumor cells in vitro [115]. Furthermore, combination of PS-targeting mAbs with immune checkpoint blockade (anti- cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or anti-PD-1) has now been evaluated in breast and melanoma syngenic models of cancer in immunocompetent mice [122, 123]. For example, Freimark et al. showed that PS-targeting enhanced the efficacy of anti-PD-1 and altered the immune landscape of tumors by increasing T-cell infiltration, proliferation and activation [123]. Taken together these data strongly suggest that the anti-cancer efficacy of PS-targeting mAb results from targeting tumor vasculature and altering the immune microenvironment of tumors by interfering with PS-mediated immune suppression (Fig. 2).

Diagram of multiple immune activation cascades upon treatment with PS-targeting mAb. Proposed pathways altered by PS-targeting mAbs that could result in improved anti-tumor immune activity
Bavituximab, a chimeric PS-targeting mAb has been evaluated in multiple clinical trials where it was found safe and well tolerated [124, 125]. Given that β2GP1 has been implicated in regulating coagulation [126] the effect of bavituximab on coagulation was evaluated closely. The phase I study saw a modest prolongation of activated partial thomboplastin timed in vitro at the highest doses given but a maximum tolerated dose of bavituximab was not identified [124]. Given the potential immune modulatory activity of bavituximab it was studied using 3D ex vivo-cultured tumor spheroids from NSCLC patients who had low PD-1 levels. Bavituximab incubation with the spheroids resulted in the increase of multiple immune-activating cytokines such as Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF), IFNγ, and TNF-α. Similar results were found in patient 3D spheroids that had low levels of PD-L1 [127]. Furthermore, Secondary analysis of a Phase III trial (SUNRISE, NCT01999673) evaluating docetaxel alone vs. docetaxel and bavituximab as a second line therapy in non-small cell lung cancer (NSCLC) showed that adding PD-1 inhibition following progression was more efficacious in patients that were treated with bavituximab + docetaxel than patients treated with docetaxel alone. Additionally, analysis of circulating cytokines demonstrated that low pretreatment serum levels of IFNγ was associated with increased efficacy with the combination bavituximab and immunotherapy [128, 129]. This suggests that PS-targeting mAbs may increase the priming T cells and highlights that the combination of PS-targeting mAbs + immune checkpoint blockade should be studied further. Ongoing trials testing bavituximab are listed in Table 5.
Conclusions
PS is an important modulator of the tumor immune microenvironment. PS-mediated immune suppression is an evolutionarily conserved pathway that tumors hijack to avoid immune surveillance. This is driven by PS interacting with PSRs, which are expressed on immune cells, endothelial cells and tumor cells. Inhibition of PSR signaling by direct targeting of PSRs or by targeting PS is currently being investigated in preclinical and clinical trials. This mini-review highlighted the contribution of TIM and TAM receptors to PS-mediated signaling in the TME; however, there are additional PSRs that induce efferocytosis and might also contribute to immune suppression. These PSRs including BAI1, CD300e, Stabilin-1 and others are worth considering in the context of anti-cancer immune therapy. Additionally, canonical signaling induced by PS is only beginning to be defined.
For instance, it is not clear if PSRs fall into classes of receptors based on signaling or cell type or potency for induction of efferocytosis and local immune suppression. We also provided an overview of the effect of PS-targeting mAbs in altering the immune landscape of tumors. While PS-targeting has advanced to clinical testing in multiple indications there are several unanswered questions that remain. The biochemical mechanism of action of the PS-targeting mAbs is yet to be fully delineated. Further it is not clear if PS-targeting mAbs interfere with all PSR signaling or a subset of PSRs. Additionally, the effect of PS-targeting mAbs on tumor cell phenotype is unexplored. This seems a potentially fruitful area of investigation given the importance of PSRs in the progression of multiple tumor types.
Understanding which patients might benefit from targeting the PS-PSR pathway is a focus for multiple groups. However, this is a challenging task given the complexity of PS-PSR biology in the TME. Highlighted by the fact that there are multiple potential sources of PS and over a dozen PSRs that might participate in PS-mediated signaling on many cell types. Thus, further research on PSRs in the context tumor immune suppression is certainly warranted.
Availability of data and materials
Not applicable.
Abbreviations
- ADC:
-
Antibody-drug conjugate
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- CAR:
-
Chimeric antigen receptor
- CEACAM-1:
-
Carcinoembryonic antigen cell adhesion molecule 1
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DC:
-
Dendritic cell
- EMT:
-
Epithelial to mesenchymal transition
- Gal-9:
-
Galectin-9
- Gas6:
-
Growth arrest-specific 6
- GM-CSF:
-
Granulocyte-Macrophage Colony Stimulating Factor
- HMGB-1:
-
High-mobility group protein box 1
- IgV:
-
Immunoglobulin domain V
- IL-10:
-
Interleukin-10
- IL-12:
-
Interleukin-12
- IL-6:
-
Interleukin-6
- INFγ:
-
Interferon gamma
- mAb:
-
Monoclonal antibodies
- MDSC:
-
Myeloid-derived suppressor cells
- NK:
-
Natural killer cell
- NSCLC:
-
Non–small-cell lung cancer
- PA:
-
Phosphatidic acid
- PC:
-
Phosphatidylcholine
- PE:
-
Phosphatidylethanolamine
- PI:
-
Phosphatidylinositol
- ProS:
-
Protein S
- PS:
-
Phosphatidylserine
- PSR:
-
Phosphatidylserine receptor
- ROS:
-
Reactive oxygen species
- RTK:
-
Receptor tyrosine kinase
- SOCS1:
-
Suppressor of cytokine signaling-1
- SOCS3:
-
Suppressor of cytokine signaling-3
- TAM:
-
Tyro3, AXL, and MerTK
- TIL:
-
Tumor-infiltrating lymphocytes
- TIM:
-
T-cell/transmembrane, immunoglobulin and mucin
- TLR:
-
Toll-like receptor
- TME:
-
Tumor microenvironment
- TNF:
-
Tumor necrosis factor
- TNFα:
-
Tumor necrosis factor alpha
- Treg:
-
Regulatory T cells
- VCAM1:
-
Vascular cell adhesion molecule 1
- VEGF:
-
Anti-vascular endothelial growth factor
- VTA:
-
Vascular targeting agent
- YW:
-
YW327.6S2
- β2GP1:
-
β2 glycoprotein 1
References
Rosenberg B, Van Camp L, Grimley EB, Thomson AJ. The inhibition of growth or cell division in Escherichia coli by different ionic species of platinum (IV) complexes. J Biol Chem. 1967;242(6):1347–52.
Rosenberg B, Vancamp L, Krigas T. Inhibition of cell division in Escherichia Coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–9.
Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene. 2012;31(39):4257–65.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23.
Hüser N, Michalski CW, Erkan M, Schuster T, Rosenberg R, Kleeff J, et al. Systematic review and meta-analysis of the role of defunctioning stoma in low rectal cancer surgery. Ann Surg. 2008;248(1):52–60.
Whitson BA, Groth SS, Duval SJ, Swanson SJ, Maddaus MA. Surgery for early-stage non-small cell lung cancer: a systematic review of the video-assisted thoracoscopic surgery versus thoracotomy approaches to lobectomy. Ann Thorac Surg. 2008;86(6):2008–16 discussion 16-8.
Green JA, Kirwan JM, Tierney JF, Symonds P, Fresco L, Collingwood M, et al. Survival and recurrence after concomitant chemotherapy and radiotherapy for cancer of the uterine cervix: a systematic review and meta-analysis. Lancet. 2001;358(9284):781–6.
Ronckers CM, Erdmann CA, Land CE. Radiation and breast cancer: a review of current evidence. Breast Cancer Res. 2005;7(1):21–32.
Barton MK. Daily aspirin may reduce mortality from prostate cancer with risk of high recurrence. CA Cancer J Clin. 2015;65(2):83–4.
Yang B, Liu T, Qu Y, Liu H, Zheng SG, Cheng B, et al. Progresses and perspectives of anti-PD-1/PD-L1 antibody therapy in head and neck cancers. Front Oncol. 2018;8:563.
Palmerini E, Picci P, Reichardt P, Downey G. Malignancy in Giant cell tumor of bone: a review of the literature. Technol Cancer Res Treat. 2019;18:1533033819840000.
Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18(1):100.
Oei SL, Thronicke A, Schad F. Mistletoe and immunomodulation: insights and implications for anticancer therapies. Evid Based Complement Alternat Med. 2019;2019:5893017.
Ritchie C, Cordova AF, Hess GT, Bassik MC, Li L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol Cell. 2019;75(2):372-81 e5. PMC6711396.
Salminen A, Kauppinen A, Kaarniranta K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): impact on cancer and aging. J Mol Med (Berl). 2019;97(8):1049-64. PMC6647228.
Ghate K, Amir E, Kuksis M, Hernandez-Barajas D, Rodriguez-Romo L, Booth CM, et al. PD-L1 expression and clinical outcomes in patients with advanced urothelial carcinoma treated with checkpoint inhibitors: a meta-analysis. Cancer Treat Rev. 2019;76:51–6.
Haddad R, Concha-Benavente F, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, et al. Nivolumab treatment beyond RECIST-defined progression in recurrent or metastatic squamous cell carcinoma of the head and neck in CheckMate 141: A subgroup analysis of a randomized phase 3 clinical trial. Cancer. 2019;125(18):3208-18. PMC6771504.
Inokuchi J, Eto M. Profile of pembrolizumab in the treatment of patients with unresectable or metastatic urothelial carcinoma. Cancer Manag Res. 2019;11:4519–28.
Kasherman L, Siu DHW, Lee KWC, Lord S, Marschner I, Lewis CR, et al. Efficacy of immune checkpoint inhibitors in older adults with advanced stage cancers: A meta-analysis. J Geriatr Oncol. 2019.
Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27.
Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5(7):551–62.
Fadeel B, Xue D. The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Crit Rev Biochem Mol Biol. 2009;44(5):264–77.
Kumar S, Birge RB. Efferocytosis. Curr Biol. 2016;26(13):R558–R9.
Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. 2018;36:489–517.
Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2013;1831(3):543–54.
Sakuragi T, Kosako H, Nagata S. Phosphorylation-mediated activation of mouse Xkr8 scramblase for phosphatidylserine exposure. Proc Natl Acad Sci U S A. 2019;116(8):2907–12.
Belzile O, Huang X, Gong J, Carlson J, Schroit AJ, Brekken RA, et al. Antibody targeting of phosphatidylserine for the detection and immunotherapy of cancer. Immunotargets Ther. 2018;7:1–14.
Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–78.
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8.
Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991;51(11):3062–6.
Koopman G, Reutelingsperger C, Kuijten G, Keehnen R, Pals S, van Oers M. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84(5):1415–20.
Doffek K, Chen X, Sugg SL, Shilyansky J. Phosphatidylserine inhibits NFkappaB and p38 MAPK activation in human monocyte derived dendritic cells. Mol Immunol. 2011;48(15–16):1771–7.
Fischer K, Voelkl S, Berger J, Andreesen R, Pomorski T, Mackensen A. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108(13):4094–101.
Martin S, Pombo I, Poncet P, David B, Arock M, Blank U. Immunologic stimulation of mast cells leads to the reversible exposure of phosphatidylserine in the absence of apoptosis. Int Arch Allergy Immunol. 2000;123(3):249–58.
Wu X, Yao Z, Zhao L, Zhang Y, Cao M, Li T, et al. Phosphatidylserine on blood cells and endothelial cells contributes to the hypercoagulable state in cirrhosis. Liver Int. 2016;36(12):1800–10.
Moller-Tank S, Maury W. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology. 2014;468–470:565–80.
Moller-Tank S, Albritton LM, Rennert PD, Maury W. Characterizing functional domains for TIM-mediated enveloped virus entry. J Virol. 2014;88(12):6702–13.
Yeung MY, Ding Q, Brooks CR, Xiao S, Workman CJ, Vignali DAA, et al. TIM-1 signaling is required for maintenance and induction of regulatory B cells. Am J Transplant. 2015;15(4):942–53.
Park D, Hochreiter-Hufford A, Ravichandran KS. The Phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19(4):346–51.
Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769–85.
Pittoni V, Isenberg D. Apoptosis and antiphospholipid antibodies. Semin Arthritis Rheum. 1998;28(3):163–78.
DeRose P, Thorpe PE, Gerber DE. Development of bavituximab, a vascular targeting agent with immune-modulating properties, for lung cancer treatment. Immunotherapy. 2011;3(8):933–44.
He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018;11:7005–9.
Ocana-Guzman R, Torre-Bouscoulet L, Sada-Ovalle I. TIM-3 regulates distinct functions in macrophages. Front Immunol. 2016;7:229.
Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–89.
Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(1):97–111.
Thomas LJ, Vitale L, O'Neill T, Dolnick RY, Wallace PK, Minderman H, et al. Development of a novel antibody-drug conjugate for the potential treatment of ovarian, lung, and renal cell carcinoma expressing TIM-1. Mol Cancer Ther. 2016;15(12):2946–54.
Wang Y, Sun J, Gao W, Song B, Shao Q, Zhao L, et al. Preoperative Tim3 expression on peripheral NK cells is correlated with pathologic TNM staging in colorectal cancer. Mol Med Rep. 2017;15(6):3810–8.
Martin-Manzo MV, Lara C, Vargas-de-Leon C, Carrero J, Queipo G, Fonseca-Sanchez M, et al. Interaction of breast cancer and insulin resistance on PD1 and TIM3 expression in peripheral blood CD8 T cells. Pathol Oncol Res. 2019;25(3):1233–43.
Liu JF, Wu L, Yang LL, Deng WW, Mao L, Wu H, et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res. 2018;37(1):44.
Hervas-Stubbs S, Soldevilla MM, Villanueva H, Mancheno U, Bendandi M, Pastor F. Identification of TIM3 2′-fluoro oligonucleotide aptamer by HT-SELEX for cancer immunotherapy. Oncotarget. 2016;7(4):4522–30.
de Mingo PA, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell. 2018;33(1):60–74 e6.
Dong J, Cheng L, Zhao M, Pan X, Feng Z, Wang D. Tim-3-expressing macrophages are functionally suppressed and expanded in oral squamous cell carcinoma due to virus-induced Gal-9 expression. Tumour Biol. 2017;39(5):1010428317701651.
Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. 2015;64(10):1593–604.
Li YM, Li Y, Yan L, Tang JT, Wu XJ, Bai YJ, et al. Assessment of serum Tim-3 and Gal-9 levels in predicting the risk of infection after kidney transplantation. Int Immunopharmacol. 2019;75:105803.
Ye L, Zhang Q, Cheng Y, Chen X, Wang G, Shi M, et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1(+) regulatory B cell expansion. J Immunother Cancer. 2018;6(1):145.
Wang Y, Zhao E, Zhang Z, Zhao G, Cao H. Association between Tim3 and Gal9 expression and gastric cancer prognosis. Oncol Rep. 2018;40(4):2115–26.
Cheng X, Li L, Thorpe PE, Yopp AC, Brekken RA, Huang X. Antibody-mediated blockade of phosphatidylserine enhances the antitumor effect of Sorafenib in hepatocellular carcinomas xenografts. Ann Surg Oncol. 2016;23(Suppl 5):583–91.
Tang D, Lotze MT. Tumor immunity times out: TIM-3 and HMGB1. Nat Immunol. 2012;13(9):808–10.
Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. 2007;26(3):311–21.
Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113(16):3821–30.
DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim Y-LE, Lee H-H, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol (Baltimore, Md : 1950). 2010;184(4):1918–30.
da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. 2014;2(5):410–22.
Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(12):1245–52.
Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81(5):1258–68.
Guo Z, Cheng D, Xia Z, Luan M, Wu L, Wang G, et al. Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer. J Transl Med. 2013;11:215.
Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. 2019;18(1):155.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94.
Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350–60.
Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv Anat Pathol. 2017;24(6):311–35.
Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–86.
Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Jia ZS, et al. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS One. 2011;6(5):e19664.
Friedlaender A, Addeo A, Banna G. New emerging targets in cancer immunotherapy: the role of TIM3. ESMO Open. 2019;4(Suppl 3):e000497.
Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119(16):3734–43.
Gleason MK, Lenvik TR, McCullar V, Felices M, O'Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. 2012;119(13):3064–72.
Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83.
Ludwig KF, Du W, Sorrelle NB, Wnuk-Lipinska K, Topalovski M, Toombs JE, et al. Small-molecule inhibition of Axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res. 2018;78(1):246–55.
Du W, Brekken RA. Does Axl have potential as a therapeutic target in pancreatic cancer? Expert Opin Ther Targets. 2018;22(11):955–66.
Williams JC, Craven RR, Earp HS, Kawula TH, Matsushima GK. TAM receptors are dispensable in the phagocytosis and killing of bacteria. Cell Immunol. 2009;259(2):128–34.
Qingxian L, Qiutang L, Qingjun L. Regulation of phagocytosis by TAM receptors and their ligands. Front Biol (Beijing). 2010;5(3):227–37.
Paolino M, Penninger JM. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers (Basel). 2016;8(10). https://doi.org/10.3390/cancers8100097. PMC5082387.
Scutera S, Fraone T, Musso T, Cappello P, Rossi S, Pierobon D, et al. Survival and migration of human dendritic cells are regulated by an IFN-alpha-inducible Axl/Gas6 pathway. J Immunol. 2009;183(5):3004–13.
O'Neill LA. TAMpering with toll-like receptor signaling. Cell. 2007;131(6):1039–41.
Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. 2013;123(8):3231–42.
Loges S, Schmidt T, Tjwa M, van Geyte K, Lievens D, Lutgens E, et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood. 2010;115(11):2264–73.
Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8(5):327–36.
Smith BN, Bhowmick NA. Role of EMT in metastasis and therapy resistance. J Clin Med. 2016;5(2):17.
Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery J-P, et al. New insights into the role of EMT in tumor immune escape. Mol Oncol. 2017;11(7):824–46.
Demarest SJ, Gardner J, Vendel MC, Ailor E, Szak S, Huang F, et al. Evaluation of Tyro3 expression, Gas6-mediated Akt phosphorylation, and the impact of anti-Tyro3 antibodies in melanoma cell lines. Biochemistry. 2013;52(18):3102–18.
Chien CW, Hou PC, Wu HC, Chang YL, Lin SC, Lin SC, et al. Targeting TYRO3 inhibits epithelial–mesenchymal transition and increases drug sensitivity in colon cancer. Oncogene. 2016;35(45):5872–81.
Shao H, Wang A, Lauffenburger D, Wells A. Tyro3-mediated phosphorylation of ACTN4 at tyrosines is FAK-dependent and decreases susceptibility to cleavage by m-Calpain. Int J Biochem Cell Biol. 2018;95:73–84.
Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang S, et al. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. 2018;17(1):20.
Kabir TD, Ganda C, Brown RM, Beveridge DJ, Richardson KL, Chaturvedi V, et al. A microRNA-7/growth arrest specific 6/TYRO3 axis regulates the growth and invasiveness of sorafenib-resistant cells in human hepatocellular carcinoma. Hepatology. 2018;67(1):216–31.
Baumann C, Ullrich A, Torka R. GAS6-expressing and self-sustaining cancer cells in 3D spheroids activate the PDK-RSK-mTOR pathway for survival and drug resistance. Mol Oncol. 2017;11(10):1430–47.
Akalu YT, Rothlin CV, Ghosh S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev. 2017;276(1):165–77.
Smart SK, Vasileiadi E, Wang X, DeRyckere D, Graham DK. The emerging role of TYRO3 as a therapeutic target in cancer. Cancers. 2018;10(12):474.
Duan Y, Luo L, Qiao C, Li X, Wang J, Liu H, et al. A novel human anti-AXL monoclonal antibody attenuates tumour cell migration. Scand J Immunol. 2019;90(2):e12777.
Sharp LL, Chang C, Frey G, Wang J, Liu H, Xing C, et al. Abstract 827: Anti-tumor efficacy of BA3011, a novel Conditionally Active Biologic (CAB) anti-AXL-ADC. Cancer Res. 2018;78(13 Supplement):827.
Ye X, Li Y, Stawicki S, Couto S, Eastham-Anderson J, Kallop D, et al. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene. 2010;29(38):5254–64.
Okimoto RA, Bivona TG. AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer (Auckl). 2015;6:27–34.
Leconet W, Chentouf M, du Manoir S, Chevalier C, Sirvent A, Ait-Arsa I, et al. Therapeutic activity of anti-AXL antibody against triple-negative breast cancer patient-derived xenografts and metastasis. Clin Cancer Res. 2017;23(11):2806–16.
Takeda S, Andreu-Agullo C, Sridhar S, Halberg N, Lorenz IC, Tavazoie S, et al. Abstract LB-277: Characterization of the anti-cancer and immunologic activity of RGX-019, a novel pre-clinical stage humanized monoclonal antibody targeting the MERTK receptor. Cancer Res. 2019;79(13 Supplement):LB-277-LB.
Cummings CT, Linger RM, Cohen RA, Sather S, Kirkpatrick GD, Davies KD, et al. Mer590, a novel monoclonal antibody targeting MER receptor tyrosine kinase, decreases colony formation and increases chemosensitivity in non-small cell lung cancer. Oncotarget. 2014;5(21):10434–45.
Du W, Huang H, Sorrelle N, Brekken RA. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight. 2018;3(21). https://doi.org/10.1172/jci.insight.124184. PMC6238734.
Kasikara C, Davra V, Calianese D, Geng K, Spires TE, Quigley M, et al. Pan-TAM tyrosine kinase inhibitor BMS-777607 enhances anti-PD-1 mAb efficacy in a murine model of triple-negative breast cancer. Cancer Res. 2019;79(10):2669–83.
Ran S, Gao B, Duffy S, Watkins L, Rote N, Thorpe PE. Infarction of solid Hodgkin's tumors in mice by antibody-directed targeting of tissue factor to tumor vasculature. Cancer Res. 1998;58(20):4646–53.
Ran S, Thorpe PE. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int J Radiat Oncol Biol Phys. 2002;54(5):1479–84.
Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62(21):6132–40.
Balasubramanian K, Schroit AJ. Characterization of phosphatidylserine-dependent beta2-glycoprotein I macrophage interactions. Implications for apoptotic cell clearance by phagocytes. J Biol Chem. 1998;273(44):29272–7.
de Groot PG, Meijers JC. Beta(2) -glycoprotein I: evolution, structure and function. J Thromb Haemost. 2011;9(7):1275–84.
Luster TA, He J, Huang X, Maiti SN, Schroit AJ, de Groot PG, et al. Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti-tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. J Biol Chem. 2006;281(40):29863–71.
Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE. Antitumor effects of a monoclonal antibody that binds anionic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res. 2005;11(4):1551–62.
Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of Docetaxel on human breast tumors in mice. Cancer Res. 2005;65(10):4408–16.
Beck AW, Luster TA, Miller AF, Holloway SE, Conner CR, Barnett CC, et al. Combination of a monoclonal anti-phosphatidylserine antibody with gemcitabine strongly inhibits the growth and metastasis of orthotopic pancreatic tumors in mice. Int J Cancer. 2006;118(10):2639–43.
He J, Yin Y, Luster TA, Watkins L, Thorpe PE. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res. 2009;15(22):6871–80.
He J, Luster TA, Thorpe PE. Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin Cancer Res. 2007;13(17):5211–8.
Mineo C, Lanier L, Jung E, Sengupta S, Ulrich V, Sacharidou A, et al. Identification of a monoclonal antibody that attenuates antiphospholipid syndrome-related pregnancy complications and thrombosis. PLoS One. 2016;11(7):e0158757.
Rahko E, Blanco G, Soini Y, Bloigu R, Jukkola A. A mutant TP53 gene status is associated with a poor prognosis and anthracycline-resistance in breast cancer patients. Eur J Cancer. 2003;39(4):447–53.
Liang Y, Besch-Williford C, Benakanakere I, Thorpe PE, Hyder SM. Targeting mutant p53 protein and the tumor vasculature: an effective combination therapy for advanced breast tumors. Breast Cancer Res Treat. 2011;125(2):407–20.
Dai B, Roife D, Kang Y, Gumin J, Rios Perez MV, Li X, et al. Preclinical evaluation of sequential combination of oncolytic adenovirus Delta-24-RGD and Phosphatidylserine-targeting antibody in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017;16(4):662–70.
Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1(4):256–68.
Gray MJ, Gong J, Hatch MM, Nguyen V, Hughes CC, Hutchins JT, et al. Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res. 2016;18(1):50.
Freimark BD, Gong J, Ye D, Gray MJ, Nguyen V, Yin S, et al. Antibody-mediated phosphatidylserine blockade enhances the antitumor responses to CTLA-4 and PD-1 antibodies in melanoma. Cancer Immunol Res. 2016;4(6):531–40.
Gerber DE, Stopeck AT, Wong L, Rosen LS, Thorpe PE, Shan JS, et al. Phase I safety and pharmacokinetic study of Bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17(21):6888–96.
Gerber DE, Spigel DR, Giorgadze D, Shtivelband M, Ponomarova OV, Shan JS, et al. Docetaxel combined with Bavituximab in previously treated, advanced nonsquamous non-small-cell lung cancer. Clin Lung Cancer. 2016;17(3):169–76.
McDonnell T, Wincup C, Buchholz I, Pericleous C, Giles I, Ripoll V, et al. The role of beta-2-glycoprotein I in health and disease associating structure with function: More than just APS. Blood Rev. 2020;39:100610. https://doi.org/10.1016/j.blre.2019.100610.
Altiok S, Valera MM, Kreahling J, Noyes D, Razabdouski TN, Kallinteris NL, et al. Activation of CD8+ tumor infiltrating lymphocytes by bavituximab in a 3D ex vivo system of lung cancer patients. J Clin Oncol. 2015;33(15_suppl):3060.
Kallinteris N, Horn L, Tang M, Guennel T, Yin S, Lai J, et al. Abstract CT159: IFN-γ analysis in blood and tissue as a potential prognostic and/or predictive biomarker. Cancer Res. 2017;77(13 Supplement):CT159–CT.
Gerber DE, Horn L, Boyer M, Sanborn R, Natale R, Palmero R, et al. Randomized phase III study of docetaxel plus bavituximab in previously treated advanced non-squamous non-small-cell lung cancer. Ann Oncol. 2018;29(7):1548–53.
Acknowledgements
We thank Dave Primm in the Department of Surgery for editorial assistance.
Funding
This work was supported by NIH grants R01 CA192381 and U54 CA210181 Project 2, the Effie Marie Cain Fellowship and a sponsored research agreement with Oncologie to RAB.
Author information
Authors and Affiliations
Contributions
ASD wrote the article which was edited by RAB. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
RAB receives research support from Oncologie, which is driving the clinical development of PS-targeting mAbs.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dayoub, A.S., Brekken, R.A. TIMs, TAMs, and PS- antibody targeting: implications for cancer immunotherapy. Cell Commun Signal 18, 29 (2020). https://doi.org/10.1186/s12964-020-0521-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-020-0521-5