
Abstract
Tyrosine kinases belong to a family of enzymes that mediate the movement of the phosphate group to tyrosine residues of target protein, thus transmitting signals from the cell surface to cytoplasmic proteins and the nucleus to regulate physiological processes. Non-receptor tyrosine kinases (NRTK) are a sub-group of tyrosine kinases, which can relay intracellular signals originating from extracellular receptor. NRTKs can regulate a huge array of cellular functions such as cell survival, division/propagation and adhesion, gene expression, immune response, etc. NRTKs exhibit considerable variability in their structural make up, having a shared kinase domain and commonly possessing many other domains such as SH2, SH3 which are protein-protein interacting domains. Recent studies show that NRTKs are mutated in several hematological malignancies, including lymphomas, leukemias and myelomas, leading to aberrant activation. It can be due to point mutations which are intragenic changes or by fusion of genes leading to chromosome translocation. Mutations that lead to constitutive kinase activity result in the formation of oncogenes, such as Abl, Fes, Src, etc. Therefore, specific kinase inhibitors have been sought after to target mutated kinases. A number of compounds have since been discovered, which have shown to inhibit the activity of NRTKs, which are remarkably well tolerated. This review covers the role of various NRTKs in the development of hematological cancers, including their deregulation, genetic alterations, aberrant activation and associated mutations. In addition, it also looks at the recent advances in the development of novel natural compounds that can target NRTKs and perhaps in combination with other forms of therapy can show great promise for the treatment of hematological malignancies.
Similar content being viewed by others

Background
Tyrosine Kinases (TKs) are a group of around 90 enzymes responsible for catalyzing the transfer of ATP phosphate group to the target protein’s tyrosine residues [1]. This substrate phosphorylation is a mechanism in which activating signals are transmitted from the cell surface to cytoplasmic proteins and the nucleus [2]. In response to external and internal stimuli, TKs have a major role in cell proliferation, survival, differentiation, and metabolism [3, 4]. Recent advancements have identified TKs role in the pathophysiology of cancer, including hematological malignancies [2, 5, 6]. Constitutive or unregulated activity and oncogenic activation in cancer cells is a common pathologic feature and can be blocked by selective TK inhibitors [4,5,6,7,8]. This is therefore considered to be a promising approach for targeted therapeutic development.
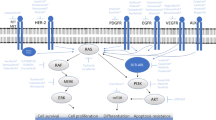
The two main classes of kinase are TKs and serine–threonine kinases (STKs) [9]. TKs are further subclassified into receptor and non-receptor proteins. Receptor tyrosine kinases (RTKs) include Platelet-derived growth factor receptors (PDGFR), Fibroblast growth factor receptor (FGFR), Epidermal growth factor receptor (EGFR), and Insulin receptor (IR). RTKs transduce extracellular signals to the cytoplasm and contain a domain that is extracellular ligand-binding, another domain that is intracellular catalytic and responsible for TK activity and regulation, as well as a disulphide bond containing transmembrane domain that connects both the ligand-binding and catalytic regions [9]. RTKs have been shown to be associated with cell division, migration and survival functions, e.g. via the phosphorylation of RAS, initiating RAF-MEK-ERK phosphorylation, consequently resulting in altered gene expression [10].
Non-receptor TKs (NRTKs) are intracellular cytoplasmic proteins that relay intracellular signals [9, 11] and can either be bound to the cell membrane or are nuclear-specific [9]. NRTKs display a broad role in cell signaling. This includes the regulation of gene expression e.g. via IL-6 mediated phosphorylation of the membrane-bound TK (Janus kinase) activating the signal transducer and activator of transcription (STAT) [12]. In addition, inhibition of cell growth e.g. via the stimulation of nuclear TKs (such as Abl) resulting in transcription factor Rb activation [13]. NRTK, such as focal adhesion kinase (FAK), can also regulate cell adhesion and proliferation [14] and are important components of signal transduction pathways, including Fyn [15] and Acks [16]. Furthermore, Acks play a vital role in cell growth via the induction of Janus kinase (JAK) and SRC machinery [17]. Tec family kinases are also associated with intracellular signaling mechanisms [18], as well as SYK which are involved in executing immune response between cell receptors and intracellular signaling [19,20,21]. Moreover, NRTKs exhibit considerable variability in their structural make up, due to a kinase domain and possession of some protein-protein interacting domains (e.g. SH2, SH3, and PH domain) [4, 22] and additional signaling. Although RTK are activated by ligand-binding, activation of NRTK involves a much more complex mode of action, incorporating heterologous protein-protein interaction, enabling transphosphorylation [23].
STKs however, similar to TKs, can be membrane-bound and nuclear. Additionally, TKs can be transmembrane receptors whereas STKs can also be cytoplasmic [9]. STKs are responsible for the phosphorylation of diverse groups of target substrates, consisting of transcription factors, regulators of the cell cycle, and cytoplasmic and nuclear effector molecules [24]. Certain growth factors, cytokines and physical or chemical induced stress collectively and/or independently act as specific triggers which regulate STK activity [25]. For instance, cytoplasmic STKs (e.g. JNK/MAPK signaling pathway) can be activated by extracellular stimuli resulting in phosphorylated JNK to be translocated to the nucleus stimulating apoptosis through JUN transcription factor [10, 26].
Research now shows that NRTKs, or members of their signaling pathways, show mutations in many forms of hematological malignant cells, which may be in fact dependent on aberrant kinase signaling for their prolonged viability and overall survival. Mutations that lead to constitutive kinase activity however have been found to result in the formation of oncogenes, including ABL, FES, Src, etc. which have been associated in the development of hematopoiesis and their function [2]. Although numerous NRTK oncogenes display differences in their structure, functionality, and subcellular localization, many exploit the same molecular pathways to enhance proliferation and viability [2].
Oncogenic NRTK mutations can be of two types, those due to point mutations, duplications or deletions and insertions, and those involving the development of a fusion gene resulting from a chromosomal rearrangement (e.g. most famously BCR-ABL). NRTK aberrant activation caused by either of these two ways is importance huge cause in the development of many hematological malignancies. Consequently, signal transduction therapy [3] and kinase inhibitors [27] have been sought after to target mutated kinases including those found to be deregulated in various hematological diseases, including lymphomas, leukemias and myelomas. A number of compounds have since been discovered which have been shown to inhibit the activity of NRTKs, which are remarkably well tolerated, considering these compounds typically target a number of kinases, including those both normal and mutant [3].
This review covers the role of various NRTKs in the development of hematological cancers, including their deregulation, genetic alterations, aberrant activation and associated mutations which give rise to such altered expression. This review further aims to showcase how the development of novel natural compounds are able to target kinases and perhaps in combination with other forms of therapy show great promise for the treatment of hematological malignancies. With particular interest in disease states associated with aggressive phenotype and the development of resistance to conventional chemotherapy, we highlight in vivo studies and clinical trials carried out targeting NRTKs with the use of natural products.
Non receptor tyrosine kinase families
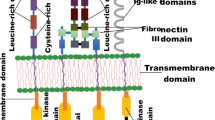
Non receptor tyrosine kinases are categorized into 9 subfamilies based on sequence similarities, primarily within the kinase domains. These includes Abl, FES, JAK, ACK, SYK, TEC, FAK, Src, and CSK family of kinases (Fig. 1).

Domain structures of various non receptor tyrosine kinase families. N: Amino terminus, SH3: SRC Homology 3 domain, SH2: SRC Homology 2 domain, Kinase: Catalytic kinase domain (also known as SH1 domain), DNA: DNA binding domain, Actin: Actin binding domain, FCH: Fes/Fer/Cdc-42-Interacting Protein homology domain, CC: Coiled coil motif, FERM: Four-point-one, ezrin, radixin, moesin domain, JH2: Janus homology domain 2 (also known as pseudokinase domain), CRIB: Cdc42/Rac-interactive domain, PH: Pleckstrin homology domain, Btk: Btk-type zinc finger motif, pr: Proline rich region, FAT: Focal-adhesion targeting domain, SH4: SRC Homology 4 domain, C: Carboxy terminus
Abl kinases
The Abelson (Abl) kinase family members include Abl1 and Abl2 (Abl-related gene, Arg), which are encoded by both ABL1 and ABL2 genes. This is one of the most conserved branches among the TKs. Human Abl1 and Abl2 proteins are ubiquitously expressed and needed for normal development. Cytoplasmic c-Abl is activated by various growth factors such as PDGF, EGFR, transforming growth factor β and angiotensin subtype 1 receptors [28]. Abl kinases links distinct extracellular stimuli to signaling cascades that regulate cell multiplication and survival, response to DNA damage and stress, dynamics of actin, cell migration, invasion and adhesion [29].
Abl1 and Abl2 kinases have a central SH3-SH2-SH1 (tyrosine kinase) domain unit, with more than 90% sequence similarity among them, and is also shared within the majority of other cytoplasmic kinases. Both have an amino terminal “cap” region and a unique long carboxy terminal tail with various protein-protein interaction sites for proteins such as p53, ATM, etc. This includes a common filamentous actin binding domain (F-BD), Abl1 specific DNA binding domain and globular domain binding with actin upstream of F-BD, and Abl2 specific second F-BD and a domain which is micro tubule binding, upstream of the F-BD. The Abl kinases have a unique cluster of three PXXP motifs, enabling interaction with other SH3 domain containing adaptor proteins such as Abi, Crk, and Nck [30]. Abl1 contains three signal motifs with nuclear localization and in the c-terminal region a nuclear export signal, which regulates its nuclear-cytoplasmic shuttling, while the Abl2 is mainly localized to F-actin rich regions within the cytoplasm and other cellular organelles owing to the lack of any nuclear localization signals [31, 32].
Abl1 was initially thought to be the oncogene vital for the generation of leukemia’s triggered by the Abelson murine leukemia virus. Later identification of the fusion oncoprotein BCR–ABL1 formed by chromosome translocation, t(9;22)(q34.1;q11.2), commonly identified as Philadelphia chromosome (Ph) confirmed the role of Abl family in cancers such as acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and acute lymphoblastic leukemia (ALL), all of which are hematological malignancies. The various malignant Abl fusion gene products encode for constitutively activated Abl kinases that can lead to cellular transformation. In solid tumors, chromosome translocation leading to activation of ABL kinases rarely exists, but is mainly regulated by its over-expression, presence of upstream oncogenic TKs or other chemokine receptors, inactivation of negative regulatory proteins and/or oxidative stress [33, 34].
Numerous intramolecular interactions influencing the SH1 kinase domain can lead to auto inhibition of the catalytic function of Abl kinases. Both SH3 and SH2 domains are involved in the regulation of auto-inhibition. Interactions amid the SH3 domain and the SH2-SH1 linker sequence as well as SH2 domain and the SH1 C-terminal lobe can lead to the formation of SH3-SH2-SH1 clamp structure, which is the auto inhibited conformation. Even a partial disruption of auto-inhibitory constraints results in oncogenic transformation. Inhibition of Abl kinases can also be achieved by interactions with lipids such as phosphatidylinositol 4,5-bisphosphate and myristoylation of amino terminal cap region. The cap region can bind intramolecularly to stabilize the inactive conformation and is required to achieve and maintain inhibition [35]. The abnormal BCR-ABL oncogenic proteins lacks auto-inhibitory cap region and the reintroduction of Abl inhibitory effects upon the reintroduction of cap region conveys the importance of the region in maintaining normal functioning.
Activation of Abl involves extensive domain rearrangements; primarily disruption of SH2 interaction with SH1 c-terminal lobe and in turn binding with the amino terminal lobe of the SH1 domain, leading to allosteric activation that is independent of the ability to bind phosphotyrosine residues. Abl activation can occur through tyrosine phosphorylation in trans by autophosphorylation, SRC family kinases induced phosphorylation and RTKs like PDGFR. Tyrosine phosphorylation of Tyr412 in Abl1 / Tyr439 in Abl2 positioned inside the activation loop of kinase domain and Tyr245 in Abl1 / Tyr272 in Abl2 located within the SH2-kinase linker domain is essential to start the kinase activity. Trans-phosphorylation of Abl1 Tyr89 located within the binding surface of SH3 domain by Src-family kinases disrupts SH3 domain–based autoinhibition leading to enhanced kinase activity and is obligatory for the full transforming activity of BCR-ABL [36]. Abl1 mediated Tyr261 phosphorylation of Abl2 increases protein stability of Abl2 [37], while phosphorylation of Tyr147 in the SH3-SH2 connector region of BCR-ABL protein by Src family kinases (Hck, Lyn, and Fyn) modulate BCR-ABL protein conformation and transforming activity [38].
Chronic myelogenous leukemia, a clonal bone marrow stem cell malignancy, is the first human cancer to be correlated with a certain genetic abnormality. CML accounts for 15% - 20% of adult leukemia’s with a frequency of 1–2 cases per 100,000 individuals. It is more common in men and is rarely seen in children. Disruption of the auto-inhibitory intramolecular interactions due chromosome translocation leads to the formation of constitutively active chimeric BCR-ABL1 fusion oncoproteins that drives CML pathogenesis.
Depending on the length of BCR sequence involved during these translocations, 3 distinct BCR-ABL variants may be created, namely p185, p210, and p230. The most common variant in CML is p210 which is observed in hematopoietic cells of CML patients in stable phase, and in ALL and AML [39]. The p230 form is associated with acute leukemia’s, neutrophilic-CML and rare cases of CML. The p185 form is found in about 20–30% of affected adults and about 3–5% of children with B-cell acute lymphocytic leukemia [40]. The loss of cap region inhibition together with the formation of a coiled-coil domain at N terminus of BCR-ABL oncoproteins causes oligomerization and subsequent proximity of numerous kinase domains leading to transphosphorylation of the critical tyrosine residues in the activation loop and other sites contributing to kinase activation [41]. BCR-ABL oncoprotein is the target of the first tyrosine kinase inhibitor (TKI), imatinib mesylate also known as STI571 which is sold under brand name of Gleevec. Majority of the FDA approved kinase inhibitors are currently in clinical use to target BCR-ABL [42]. Imatinib mesylate is an orally available ATP-competitive inhibitor that works by stabilizing the inactive ABL kinase–domain conformation. Nilotinib, Dasatinib, Bosutinib and Ponatinib are second generation TKI used for imatinib mesylate resistant cases.
While BCR-ABL is the most common chromosomal translocation, several other chromosomal abnormalities lead to the expression of various fusion proteins, but there are no activating point mutations identified in the ABL1/ABL2 genes. Various Abl1 fusion proteins involved in hematological malignancies includes BCR-ABL1 (p210), BCR-ABL1 (p185), BCR-ABL1 (p230), NUP214-ABL1, EML1-ABL1, ETV6-ABL1, ZMIZ1-ABL1, RCSD1-ABL1, SFPQ-ABL1, FOXP1-ABL1, SNX2-ABL1, RANBP2-ABL1; while ETV6-ABL2, RCSD1-ABL2, PAG1-ABL2 and ZC3HAV1-ABL2 are originating from Abl2. A large number of signaling pathways are activated by BCR-ABL, but the pathways that are critical for BCR-ABL–dependent transformation includes Gab2, Myc, CrkL and STAT5 [43].
Presence of BCR-ABL oncoprotein is the most frequent genetic abnormality found in adult ALL patients. Nearly 3–5% childhood and 25–40% adult cases of ALL have Philadelphia chromosome, the presence of which confers a worst prognosis and most of these cases present with an aggressive leukemia. First generation tyrosine kinase inhibitor, imatinib mesylate monotherapy can lead to complete remission rates (90%–100%) and combining imatinib mesylate with standard chemotherapy also increases the overall long-term disease-free survival in both adults and children. Imatinib mesylate based induction and consolidation regimens followed by hematopoietic stem cell transplantation significantly improved the outcome Ph+ ALL [44].
Approximately 1% of newly diagnosed AML cases show a consistent association with the Ph chromosome [45]. The presentation of cases with CML in myeloid blast crisis and Ph+ AML needs stringent criteria to differentiate. Characteristic of Ph+ AML includes co-occurrence of typical metaphase chromosome alongside Ph+ metaphases during diagnosis, less probability for additional copies of Ph and trisomy 8. Ph+ AML patients will have a poor prognosis with standard chemotherapy regimen, and would benefit from combination therapy with imatinib mesylate [46].
Feline Sarcoma (FES) kinases
FEline Sarcoma (FES) and FEs Related (FER) are members of a separate class of NRTKs called FES kinase family. These kinases are homologous to the viral oncogenes; feline v-fes (Feline sarcoma) and avian v-fps (Fujinami poultry sarcoma) which are responsible for cancerous transformation. Fes, a 93KDa proto-oncogene, is predominantly present in the myeloid lineage of hematopoietic cells, epithelial, neuronal and vascular endothelial cells, while Fer is ubiquitously expressed. Human c-Fes has been linked to multiple cell surface growth factor and cytokine receptors (ex, interleukin 3 & 4 and GM-CSF receptors) which are involved in cell survival and migration, release of inflammatory mediator and innate immune responses. In addition, it might play a direct part in myeloid differentiation and angiogenesis [47].
Recent findings show that both kinases remain activated in primary AML blasts as well as cell lines. Fes has been reported to have a role in phosphorylation/activation of STAT family of transcription factors, and signaling proteins such as phosphatidylinositol-4,5-bisphosphate 3-kinase, mitogen-activated protein kinases and extracellular signal–regulated kinases [48]. Fes is essential for downstream signaling of the mutated oncogenic KIT receptor. Both Fes and Fer are involved in regulation of vital functions downstream of internal tandem duplication containing FLT3. Fer kinase is necessary for cell cycle progression, while Fes is needed for D816V mutated KIT dependent cell survival.
FES kinases have a unique amino terminal FCH (Fes/Fer/Cdc-42-interacting protein homology) domain, three coiled-coil motifs that facilitate oligomerization, a central SH2 domain for various protein-protein interactions and a kinase domain in the carboxy terminal region. FCH domain together with the first coiled-coil motif is called F-BAR (FCH-Bin–Amphiphysin–Rvs) domain [49]. The biological activity of Fes is tightly regulated, with a tight packing between SH2 and kinase domain to maintain a catalytically repressed state, so that kinase activity is regulated despite the absence of a negative regulatory SH3 domain. Activation of Fes kinase needs active phosphorylation of Tyr713 located inside the activation loop. Tyr 811 is another critical phosphorylation site for the activation of Fes.
Aberrant activation of Fes is not connected with human cancers. Regardless, studies show that hyper-activation of Fes kinase is critical in maintaining the deregulated proliferation in human lymphoid malignancies elicited by constitutively active forms of mutated surface receptors (internal tandem duplication containing FLT3 and KIT D816V) [50]. Four somatic mutations within the kinase domain of Fes was reported in colorectal cancers, but none of them are gain-of-function mutations [51]. Similarly Fer mutations in small cell lung cancer has been reported [52] Over expression of human c-fps/fes using retroviral vector can transform fibroblasts and other established mouse cells [53] and it requires Ras, Rac, and Cdc42 [47].
JAK kinases
JAK family of tyrosine kinases consist of four members that includes JAK1, JAK2, JAK3, and Tyk2 [54]. All members of JAKs family contain a similar protein structure; a carboxy terminal kinase domain flanked by a catalytically inactive JH2 (Janus homology domain 2), pseudokinase domain which possesses a kinase-regulatory activity via a SH2 domain. There is also a FERM domain that regulates the binding to the membrane-proximal part of the cytokine receptors [55, 56]. Following binding of the ligand (usually cytokines, such as interferon α/β/γ, interleukins, GPCR ligands and growth factors) to specific receptor, these kinases are activated [57] via tyrosine phosphorylation of the cytoplasmic domains of cytokine receptors [58]. Activated JAKs then subsequently phosphorylate cytoplasmic domain of the receptor [59]. The resulting complex of receptor then recruits and phosphorylates the cytoplasmic STAT family members [60, 61]. STAT family members are major downstream targets of JAK kinases in the pathogenesis oh hematological malignancies [62]. STAT phosphorylation is followed by dimerization and translocation from cytoplasm to the nucleus, where it regulates the manifestation of various target genes [54, 63].
Constitutive activation of JAKs has been reported in many cancers including various hematological malignancies. Deregulated JAK activity arises by numerous means, including aberrant cytokine production via autocrine/paracrine mechanism, activating point mutations within JAKs or any other oncogene upstream of signaling cascade.
Through past several years, a number of JAK mutations that lead to activation of constitutively active or hyperactive JAK activity have been identified [64]. The genetic alteration of JAK family has been reported in all members. It is a well known fact that JAK mutations is linked with development of hematological malignancies [59, 65]. The majority of these alterations are point mutations [59]. JAK2V617F mutation is one of the most studies genetic alteration in JAK family [59]. JAK2V617F mutation is mainly found in primary myelofibrosis or essential thrombocythemia patients. These patients have an incidence of 50% to 60% JAK2V617F mutational freqency and majority (95%) reported polycythemia vera [66]. Another point JAK1 mutation, A634D has been reported in the pseudokinase domain [67]. This mutation has been shown to cause a prominent effect on signaling functions [67]. JAK1 mutation has been found to involve in the development of AML [68] JAK1 mutations are commonly found in T-cell ALL (18%) and with a lesser frequency in B-cell ALL (B-ALL). Constitutive activation of STAT5 has been linked with mutation of JAK1 [65, 69, 70]. JAK1 mutation-mediated activation of STAT5 is also reported in AML patients. JAK3 member of JAK family is found only in hematopoietic lineage. Point mutations leading to aberrant activation of JAK3 have been reported in various leukemia/lymphomas [71]. Juvenile myelomonocytic leukemia (JMML) patients with secondary mutations in JAK3 have poor prognosis and clinical outcome. In JMML 12% of JAK3 gene has been found to be mutated [72]. Mutation of JAK3 is reported in 15% acute megakaryoblastic leukemia [73]. T-cell lymphoma patients (Extranodal nasal-type natural killer) (21%) were reported to have JAK3 mutations (A573V or V722I) in the pseudokinase domain [74]. These mutations can lead to constitutive JAK3 activation conferring invasive growth and survival advantages. In aggressive T-ALL, JAK3 mutation has been found to be significantly associated [75]. Mutation inTYK2 kinase have been reported in T-ALL (21%) and play a role in promote cell survival via activation of STAT1 as well BCL2 upregulation expression [76].
JAK2 amplification via telomeric segment translocation (9p24) leading to increased JAK2 expression and kinase activity has been described in Hodgkin lymphoma and primary mediastinal B-cell lymphoma [77,78,79].
ACK kinases
Acks also known as Activated Cdc42 kinases (Acks) are the important components of signal transduction pathways which comes under the category of non-receptor tyrosine kinases. There are seven different types of Acks viz., Ack1/Tnk2, Ack2, DACK, TNK1, ARK1, DPR2 and Kos1 [16]. Most of the members of the Acks are evolutionary conserved and consists of both N-terminal and C-terminal domains such as a SH3 domain and a kinase domain with key difference in the protein’s c-terminal region [16, 80]. Presence of C terminal kinase domain followed by a SH3 domain along with (CRIB) makes them unique NTRKs [16, 80].
Ack1 (ACK, TNK2, or activated Cdc42 kinase) is one of the most widely studied and first well known members of the Acks. Ack1, a ubiquitous 140KDa protein located on the chromosome 3q, was first cloned in hippocampus of the human brain that binds to active form of CdC42 i.e., in its GTP bound form [80, 81]. Presence of multiple structural domains (N-terminal; SAM domain, tyrosine kinase catalytic domain, SH3 domain, CRIB domain, and C terminal; proline rich domain, ubiquitin associated domain) makes ACK1 distinct from other NRTKs and also provides strong force for its functional diverseness [16, 82].
ACK1 play vital role in cell survival, migration, cell growth and proliferation via acting as an integral cytosolic signal transducer for the array of receptor tyrosine kinases (MERTK, EGFR, PDGFR, IR etc.) to different intracellular effectors which includes both cytosolic as well as nuclear [81]. Furthermore, Ack1 is also an important epigenetic regulator with negative regulatory effect on tumor suppressors [81,82,83,84,85,86].
Considerable number of reports has revealed crucial role of ACK in the carcinogenesis of various types of neoplasm. Abnormal overexpression, amplification, or mutation of ACK1 has been well documented in many forms of human cancers, including gastric, breast, ovarian, pancratic, colorectal, head and neck squamous cell carcinomas, osteosarcoma, hepatocellular carcinoma, and prostate cancers [81, 85,86,87,88,89,90]. Recently, Xu et al., revealed that ACK1 promotes development of gastric tumors by p53 ubiquitination degradation via upregulating ecdysoneless homolog, a cell cycle regulator [86] and also reported earlier that ACK regulates the expression of about 147 proteins which are closely associated with cell survival [91].
A number of underlying mechanisms have been documented for ACK1 mediated cancer development. Recently, Maxon et al. reported that mutations in ACK1/TNK2 gene is the main oncogenic cause for AML and chronic myelomonocytic leukemia and that these mutations were sensitive to inhibitors of ACK1 [92]. Furthermore, in the case of chronic neutrophilic leukemia and atypical CML, ACK1 plays critical role in growth by inducing JAK and SRC machinery [17]. In acute leukemia patients harboring NRAS mutation, ACK1 along with other survival proteins have been identified as important therapeutic targets [93]. Diverse crucial role of ACK1 implicated in carcinogenesis including the stimulatory effect on the array of signaling molecules related to cancer development such as AKT, AR, and also by down regulation of tumor suppressors entails its therapeutic importance and prompts the community to look for potential inhibitors.
TNK1 (thirty-eight-negative kinase 1), another important member of ACK family NRTKs of size about 72 KDa, was first reported in blood stem cells of human umbilical cord and murine embryonic cells [16, 94]. Available literature reveals that TNK1 has both tumor suppressing and oncogenic potential as it can mitigate the growth of tumor cells by dowenregulating Ras-Raf1-MAPK pathway [95], induce apoptosis through NF-κB inhibition [96], activate cellular transformation and growth of neoplastic cells [97, 98]. TNK1 sorted out as an important kinase with oncogenic potential implicated in hematological carcinogenesis such as in AML and Hodgkin’s Lymphoma which suggests that targeted intervention of TNK1 may open new platform for therapy.
SYK kinases
Spleen tyrosine kinase (SYK) is one of the important members of soluble non-receptor protein kinases of syk family and was first cloned in procrine spleen cells with reported highest expression cells of hematopoietic origin [99, 100]. It is a 72 kDa protein encoded by SYK gene located on chromosome 9q22 and consists of highly conserved two SH2 domains with N-terminal and one tyrosine kinase domain at C-terminal and is highest homologous to ZAP-70 [19, 20, 100,101,102]. SYK is activated by C-type lectins and integrins, and the downstream signaling cascade includes VAV family members, phospholipase Cγ isoforms, the regulatory subunits of phosphoinositide 3-kinases and the SH2 domain-containing leukocyte protein family members (SLP76 and SLP65) [20].
SYK as cytosolic NRTK have crucial role in immune response between cell receptors and intracellular signaling machinery via phosphorylating cytosolic domain of the immunoreceptor tyrosine-based activation motifs (ITAMs) that result in the conformational changes and further activation of SYK which then transduces signal to other downstream target/effector proteins [19,20,21]. Various findings documented the critical role of SYK in many forms of hematological malignancies by virtue of its stimulatory effect on various survival pathways/signaling molecules [103,104,105]. SYK has also been found to have tumor suppressive effect in the cancers of non immune cells [106]. Considering important role of SYK in the development of malignancies, progress can be made in the development of effective anticancer molecules.
TEC kinases
Tec family kinases, the second largest subfamily of the NRTKs, consist of five members, including BTK (Bruton’s tyrosine kinase), ITK/EMT/TSK (interleukin 2-inducible T-cell kinase), RLK/TXK (tyrosine-protein kinase), BMX/ETK (bone marrow tyrosine kinase on chromosome) and Tec (tyrosine kinase expressed in hepatocellular carcinoma) [107]. One of the main features of Tec is the presence of an amino terminal pleckstrin homology (PH) and Btk-type zinc finger (BTK) motif followed by a SH3 and SH2 domains and a carboxy terminal kinase domain in their protein structure. As PH domain can bind phosphoinositides, Tec family kinases are assumed to act as the connection between phosphotyrosine-mediated and phospholipid-mediated signaling pathways. Tec kinases are associated with cellular signaling pathways of cytokine receptors, RTKs, lymphocyte surface antigens, G-protein-coupled receptors and integrins [18]. Tec are abundantly expressed in hematopoietic cells and contribute towards their growth and differentiation [18].
Mutations found in the gene BTK, essential for B-lymphocyte development, differentiation, and signaling [108], have been shown to trigger the human B-cell immunodeficiency, X-linked agammaglobulinemia and X chromosome-linked immunodeficiency in mice. This not only proved that BTK activity is required for B-cell development, but supports the presumption that Tec family proteins are crucial for growth and maturation of blood cells [18]. Previously, the majority of indolent B-cell lymphoma patients did not enter complete remission with treatment and inevitably relapsed [109]. Over the past 10 years, innovative immunochemotherapies have increasingly improved disease control rates but not survival. Therefore, the development of novel agents were urgently needed, which targeted dysregulated pathways in hematological malignancies. In addition, recent preclinical data has illustrated that BTK is present in specific tumor subtypes and in other relevant cells contributing to the tumor microenvironment, e.g. dendritic cells, macrophages, myeloid derived suppressor cells and endothelial cells. BTK inhibitors against hematological malignancies [110] have hence been developed, most notably Ibrutinib (PCI-32765), a first in class covalent inhibitor of BTK. Ibrutinib is an orally available small molecule approved for the treatment of patients with some hematological malignancies and It has been proposed that Ibrutinib may also display antitumor activity in solid neoplasms [111]. Ibrutinib is claimed to be a “breakthrough therapy” by the FDA [109] and overall has changed the future outlook of therapy for lymphoma.
ITKs, the predominant and highly expressed Tec kinase in T cells, act as vital signaling mediators in normal as well as malignant T-cells and natural killer cells [112]. Thus, playing an important role in autoimmune inflammatory diseases [113]. ITK is involved in a variety of downstream signaling from T-cell and NK cell surface receptors and RTKs, mainly the T-cell receptor and Fc receptor [114,115,116]. ITK mediates signaling by activating phospholipase Cγ1, resulting in activation of downstream targets such as, nuclear factor of activated T-cells (NFAT), NFκB, and mitogen-activated protein kinase pathway [117]. ITK inhibitors could hence have therapeutic potential in several autoimmune, inflammatory, and malignant diseases. For example, in a recent study by Zhong et al. [112], using the novel ITK/RLK inhibitor PRN694, ex vivo assays reported inhibitory activity against T-cell prolymphocytic leukemia cells.
TXK expression is mainly detected in some myeloid cell lines and T-cells. Moreover, TXK is expressed in T-cell subsets (e.g. Th1/Th0 cells), and was reported to act as a Th1 cell-specific transcription factor, regulating IFN-γ gene expression via binding to its promoter region, increasing transcriptional activity [118]. An increasing amount of interest has focused on T-cell subsets, which have been characterized, based on their array of cytokine production, e.g. Th1 cells have been found to secrete IL-2, IFN-γ, and lymphotoxin, supporting cell-mediated response [118,119,120,121,122].
BTK, ITK, and TXK have shown selective expression in bone marrow cells [123]; however BMX and TEC have displayed a much broader expression, even extending to normal somatic cells (e.g. cardiac endothelium) [107]. BMX has been reported to be expressed in myeloid lineage hematopoietic cells (e.g. granulocytes and monocytes), endothelial cells, and numerous types of oncologic disorders [107]. Over the past decade, there was significant progress made in this area of research, which has suggested a prominent role of BMX in cell survival, differentiation and motility, and as such, a key player in inflammation and cancer [107, 124].
TEC is expressed in hematopoietic cells like myeloid lineage cells, B and T cells, as well as neutrophils and has been reported to be involved in the stabilization of lymphocytes (B and T), T and B cell receptor signaling, and in the nuclear factor activation of activated T-cells [125]. The overexpression of TEC has been found to be associated with tumorigenesis and liver cancer progression [126]. Inhibiting TEC or degrading the phosphorylation of TEC may therefore have a direct affect on the progression and development of liver cancer. This was supported by an investigation carried out by Chen et al. [127] exploring TEC protein expression in hepatocellular carcinoma and TEC phosphorylation in 200 specimens of cancerous and non-cancerous liver tissue. A more recent study by Vanova et al. [128] with interest in the expression of TEC in hepatocellcular carcinoma, identified TEC as a regulator in controlling pluripotent cell fate in human pluripotent stem cells, acting through the regulation of fibroblast growth factor-2 secretion. Such studies provide further support and evidence of the broad activities and roles of tyrosine kinase preferentially expressed in hepatocellular carcinoma.
Focal adhesion kinase
FAK family consists of 2 members; the ubiquitously expressed focal adhesion kinase, and the associated adhesion focal tyrosine kinase (Pyk2) that is manifested in the central nervous system and in hematopoietic cells. FAKs play a part in the normalization of cell propagation and adhesion as well as cell to microenvironment communications [14]. FAK over expression has been reported in B-lymphoblastic leukemia and lymphoma cells. Interestingly, FAKs are absent in leukemias/lymphomas of T-cell origin as well as in myeloma [129]. FAK responds to extracellular stimuli, including signals from the extracellular matrix to regulate cell proliferation and migration [130]. Interaction of growth factor with receptor has been found to activate leading to phosphorylation/activation of Src kinase. The activated Src kinase then, via association with several signaling pathways, regulates cell proliferation and survival of cancer cells [131]. FAK expression is found to be significantly higher in MDS patients with CD34+ and CD34+ CD38− as compared to patents with normal CD34+ [131]. FAK overexpression has been linked to the enhancement of leukemic cell migration from the marrow to the circulating compartment, associated with drug resistance [132]. FAK regulate cell migration through regulation of Rho family of proteins, paxillin kinase linker (PKL/Git2)–β-pix complex and phosphatidylinositol 4,5-bisphosphate. FAK overexpression in AML has been found to be associated with poor survival outcome [132, 133].
Src kinases
The Src family of tyrosine kinases (SFKs) are membrane-associated NRTKs active as key mediators of biological signal transduction pathways. This family includes 11 related kinases: Blk, Fgr, Fyn, Hck, Lck, Lyn, c-Src, c-Yes, Yrk, Frk (also known as Rak) and Srm [134, 135].
SFK members share a highly conserved structure, comprising of a membrane-targeting myristoylated or palmitoylated SH4 domain in the amino terminal region, trailed by SH3, SH2 and a kinase domains, and a short carboxy terminal tail with an auto-inhibitory phosphorylation site [134]. Furthermore, each member of SFKs has a specific domain of 50–70 residues that is consecutive to the SH4 region and divergent among the different family members [136].
Five members of the SKFs (Blk, Fgr, Hck, Lck and Lyn) are expressed predominantly in hematopoietic cells. However, c-Src, c-Yes, Yrk and Fyn, are expressed ubiquitously with high levels in platelets, neurons and some epithelial tissues [134, 137]. Moreover, Srm is present in keratinocytes, and Frk expressed primarily in bladder, breast, brain, colon, and lymphoid cells [135].
SFKs have a major role in a variety of cellular signaling pathways activated through various RTKs (PDGF-R, EGF-R, FGF-R, IGF1-R, CSF-R) [138] and G-protein coupled receptors, regulating cell survival, DNA synthesis and division, actin cytoskeleton rearrangements and motility [137, 139]. Src family member exerts its full catalytic activity upon phosphorylation of a critical residue (Tyr419) within the activation loop. Phosphorylation of the auto-inhibitory site Tyr530 within the carboxy terminal tail forms a closed auto-inhibited inactive conformation via the association of the SH2, SH3, and kinase domains by intramolecular interactions. Many factors, including specific cellular signals, or transforming mutations, could break these interactions and produce an active open kinase [140]. Several protein tyrosine phosphatases can dephosphorylate Tyr530 and thus regulate its kinase activity.
SFKs associate with PDGF-R via an interaction of their SH2 domain with Tyr579 of the ligand bound activated receptor. This association will release the auto-inhibitory intra-molecular interface between the SH2 domain and the carboxy terminal tail, subsequently permitting the formation of catalytically active conformation. SFKs in turn modulate RTK activation and are involved in promoting mitogenesis.
SFKs might have a part in cancer development due to their implication in the regulation of cell–cell adhesion. This regulatory pathway involves different molecules such as p120-catenin protein, a substrate of Src [141]. SFK, particularly Src, might also be involved in tumorigenesis by activation of STAT transcription factors which regulate cytokine signaling in hematopoietic cells [142]. Moreover, SFKs like focal adhesion kinase, paxillin and p130CAS have been implicated in monitoring of signaling pathways mediated by integrin. Alterations in integrin activity have been described in several tumor types [143]. Src is also thought to have a role in the progression of CML, AML, CLL, and ALL through activation of STAT pathways and regulation of RAS/RAF/MEK/ERK MAPK and VEGF pathways. Other oncogenic pathways of C-Src include translocation of B-catenin, promotion of ERK and Cbl phosphorylation and increase in anti-apoptotic Bcl2 in cancer cells [144,145,146].
SFKs also play a role in the development and signaling of T and B cells. Indeed, SFKs, particularly Lck, appear necessary for T cell receptor-based signaling essential for various phases of T-cell development [134, 147]. In addition, Lyn, have a major role in B-cell lineage development and maturation, activation as well as inhibition [148].
A consistent number of studies point the role of SFKs in human tumors since they are often overexpressed and/or constitutively hyper-activated in several cancers [137]. Activation of SFKs could arise either after a mutation of Src allele leading to disrupted negative regulatory network or to binding of SFKs to activating partners such as growth factor receptors (Her2/Neu, PDGF, EGFR, c-kit), adaptor proteins and other NRTKs (focal adhesion kinase, Bcr-Abl) [149]. Various SFKs members have been implicated in the development of hematopoietic malignancies such as leukemia and lymphomas (AML, ALL, CML, Burkitt’s lymphoma, etc.) [150]. However, oncogenic mutations of SFKs are rarely observed in hematological malignancies [151]. Therefore, the progression of leukemia and lymphoma malignancies is mainly associated with the constitutive activation of SFKs and to amplification of anti-apoptotic and oncogenic downstream signaling pathways [149, 150].
In cancer cells, multiple mechanisms are able to disrupt the inactive conformation of SFKs including binding of SH2 to activated receptors such as flt3 (in AML) and to oncogenic protein kinase such as BCR-ABL (in CML and ALL) [152]. Furthermore, in cancer cells, the SFKs inhibitory signaling pathways such as C-terminal Src kinase have shown to be suppressed thus leading to a stabilization of SFK activated conformation [151]. Activation of SFKs promotes multiple downstream signal transduction cascades implicated in apoptosis and oncogenesis (STAT3 and STAT5, MEPK, EGFR, PDGFR, PI3K/AKT and VEGFR) [146, 149, 150, 153].
Moreover, it has been shown that SFKs promote cancer cells resistance to chemotherapy and radiation as well as targeted RTK therapies [154, 155]. Donato et al. have demonstrated that Lyn and Hck, were upregulated in imatinib mesylate resistant cell line and in specimens of advanced CML and ALL from patients who relapsed to imatinib mesylate [149, 156]. Indeed, SFKs members, particularly Hck and Lyn, interact with the oncogenic BCR-ABL fusion protein and promote resistance to imatinib mesylate treatment [157].
Given the importance of SFKs in multiple aspects of tumor development, such as proliferation, migration, resistance to apoptosis, and angiogenesis, these proteins can be considered as attractive targets for future anti-cancer therapeutics. Moreover, inhibition of SFKs in combination with standard anti-cancer therapies has been suggested as a promising treatment strategy with a clinical potential in overcoming resistance to current regimens and preventing metastatic recurrence [154].
The viral encoded Src (v-Src) is constitutively active and highly transforming, where as c-Src over expression does not induce transformation. v-Src transformed cells, but not c-Src over expressing cells, have the ability to form tumors in nude mice [158]. But mutant form of c-Src created by single amino acid changes (Thr to Ile at position 338/Glu to Gly at position 378/Phe to Ile at position 441) or by fragment of c-src (Gly-63, Arg-95, and Thr-96) with a corresponding fragment of v-src (Asp-63, Trp-95, and Ile-96) is oncogenic and induce transformation ([159, 160].
Fyn has been found to be overexpressed in various types of cancers including hematological malignancies [15, 161, 162]. Fyn is involved in development and activation of T cells [15]. Activated Fyn is proven to play a role in the pathogenesis of multiple human carcinomas via influencing cell growth, transformation ability of cells and apoptosis [15]. Fyn has been also found to participate in generation of mitogenic signaling, initiation of cell cycle and cell to cell adhesion [163]. Fyn also play a critical role in aggressiveness of CLL.
Lyn is aberrantly expressed and highly activated in many cancer cells [164, 165]. Association of Lyn kinase with dysregulated signaling pathways in various hematopoietic as well solid tumors implicates that it might be an important target for the treatment of cancer. Dysregulation of Lyn have an important role in progression of CLL via regulation of apoptotic signaling pathway [166]. A number of substrates has been identified in CLL including SYK, PI3K, HS1, procaspase-8, and PP2A [167,168,169].
C-terminal Src kinases
C-terminal Src kinases (Csk) and Csk-homologous kinase (Chk) are the two members of this family of NRTKs. Csk is a 50 kDa protein with an amino terminal SH3 domain followed by a SH2 domain and a carboxy terminal kinase domain. A characteristic feature of Csk is the absence of a site in the activation loop for auto-phosphorylation. The active conformation is stabilized by the binding of SH2-kinase and SH2-SH3 linkers to the amino terminal lobe of the kinase domain.
CSKs phosphorylates the auto-inhibitory tyrosine residues in the Src-family kinases’s C-terminal tail which stabilizes SFKs in a closed inactive conformation and thus functions as the major endogenous negative regulators of SFKs. Chk can engage a complementary mechanism to inhibit SFKs by direct binding to SFKs, which is also called as non-catalytic inhibitory mechanism. Several other signaling proteins such as paxillin, P2X3 receptor, c-Jun and Lats can also serves as substrates of Csk, but the physiological relevance of it is not yet known [151, 170].
Csk is ubiquitously expressed in all cells, however, Chk is mainly expressed in the brain, haematopoietic cells, colon tissue and smooth muscle cells [170]. Csk is primarily present in cytosol as it does not have a transmembrane domain or any fatty acyl modifications. As the substrate molecules (SFKs) are attached to the membrane, the mobility of Csk to the membrane by means of numerous scaffolding proteins (caveolin-1, paxillin, Dab2, VE-cadherin, IGF-1R, IR, LIME, and SIT1), is a crucial step in the regulation of Csk activity [151].
They have an important role in the regulation of cell functions like growth, migration, differentiation, and immune response. Recent studies suggest that Csk can have a function as tumor suppressor through the inhibition of SFKs oncogenic activity.
Targeting NRTKs using Natural products
Past few years have seen tremendous improvement in the area of drug discovery in field of cancer. Although many new entities are available in market for therapeutic treatment but association of adverse events like acute/chronic organ damage, suppression of bone marrow, and potential toxicities like hepatic, renal, gastrointestinal etc., with these drugs limits its use [171,172,173,174] continuing quest for search of newer and effective molecule.
Nowadays targeted therapy are gaining high importance due to its ability to directly act on specific molecule and signaling pathway. Tyrosine kinases compete with ATP binding site of the catalytic domain of oncogenic tyrosine kinases and modulate signaling pathway [175]. Thus it becomes very important to target such kinases by using specific drugs that aims directly at kinases.
Inhibitors like IFN-alpha regulates T-cells however, due to patient non-compliance and side effects produced thereby limited its use. Second generation tyrosine kinase inhibitors like dasatinib, nilotinib, bosutinib along with imatinib mesylate have gained tremendous respect as a conventional chemotherapeutic agent for treatment in CML patients. Despite of great achievements in the therapeutic treatments of CML, search for newer effective and potent agent toward resistant mutants such as T315I is continued. Drugs like aurora kinases, ponatinib were effective against resistant mutant but due to cardiac toxicity and maximum tolerated dose for ponatinib being 45 mg their use were limited [5, 175,176,177,178,179,180,181]. Natural products are now considered as an alternative for synthetic drugs.
Secondary metabolite/s present in natural products is known to possess diverse biological effects. These natural products are present in numerous sources like plants, micro-organism, fungi etc. Besides being non-toxic in nature they are considered to less expensive. In 2013, US FDA approved 1453 new chemical entities from which 40% comprised of natural products or analogs of natural compounds [182, 183]. Natural products alone or in combination have been able to induce apoptosis as well chemosensitised those cell lines that were resistant to conventional drugs. Below we have discussed compounds (Fig. 2) that hold a high potential to be developed as a lead molecule as a tyrokinase inhibitor. Some of these natural compounds has in vivo (Table 1) data and some has progressed through clinical trials (Table 2).

Chemical structures of various natural products targeting non receptor tyrosine kinases
Omacetaxine mepesuccinate
Omacetaxine mepesuccinate is also known as homoharingtonine (HHT) (Fig. 2a) is a cephalotaxine ester isolated from bark of Cephalotaxus species and belongs to class of alkaloidal compounds. Documented reports claim that practioners from Fujian Province of China used the extract of HHT in treatment of cancer. However limited availability of this drug led to development of semisynthetic compounds with better efficacy and safety compared to parent drug. It is only one such natural drug that owes US FDA approval for treatment of CML patients that developed resistant or failed to respond to conventional chemotherapeutic tyrokinase inhibitor. The liver is able to metabolize Omacetaxine mepesuccinate without causing liver toxicity and have biphasic half life of alpha 0.5 ± 0.1 h and beta half life of 9.3 ± 1.4 h [184, 185].
Omacetaxine has been studied using cell lines (BCR-ABL- expressing myeloid and lymphoid) and in in vivo mouse model of CML and B-cell acute lymphoblastic leukemia bearing BCR-ABL or BCR-ABL-T3151 mutation. Both in vitro and animal model results demonstrated reduction in number of leukemic cells in both CML and in mice models. Besides this, omacetaxine also repressed expression of BCR-ABL-T3151 expressing leukemic cells [186, 187]. Similar effects were demonstrated in study comprising of omacetaxine mepesuccinate and nilotinib in combination [188]. However, published data specifies that omacetaxine does not rely on BCR-ABL binding site to exhibit its activity instead it blocks synthesis of protein by competing with amino acid side chain of aminoacyl-tRNAs to bind with A-Site cleft of ribosomal unit. [189, 190].
In the early 1980s omacetaxine mepesuccinate was used in the treatment of CML patients. In clinical trial (phase I/II), six patients who did not respond to conventional chemotherapeutic tyrosine kinase inhibitor imatinib mesylate, responded to omacetaxine mepesuccinate treatment. This evaluable effect was observed in five patients. Complete blood related response was observed in all patients whereas three of them showed genetic response at cellular levels. Efficacy of treatment was measured on basis of decreasing levels of BCR-ABL transcripts [191].
Cytogenetic response along with absence of BCR-ABL mutation was witnessed in two patients harboring BCR-ABL kinase mutation before start of omacetaxine treatment [192]. Efficiency of omacetaxine mepesuccinate was studied in Phase II clinical trial consisting of patients bearing BCR-ABL-T1351 mutation. Median rate of seven cycles of omacetaxine mepesuccinate treatment were received by 62 patients. Out of 62, 48 patients showed signs of complete hematologic response whereas, 14 patients achieved major cytogenetic response. Progression free survival rate was reported to be for 7.7 month [193].
So far couples of phase II clinical studies have verified effect of HHT and omacetaxine mepesuccinate in patients with CML who were in early stage of the disease or in the late chronic phase. In all 212 CML patients received either HHT or omacetaxine mepesuccinate at dose of 2.5 mg/m2 for 14 days or 1.25 mg/m2 twice a day for 14 days respectively. Complete hematologic response rate was 80% whereas cytogenetic rate was found to be 42% [190, 192,193,194]. In another clinical study consisting of 252 patients who did not respond or developed resistance to two or more tyrosine kinase inhibitor were treated with omacetaxine mepesuccinate at a dose of 1.25 mg/m2 twice a day for 28 days and then less than 7 days/cycle as maintenance dose showed 20% of cytogenetic response [195, 196].
Omacetaxine mepesuccinate was also tried in tandem with other treatment agents and drugs. Efficiency of omacetaxine mepesuccinate was tested in combination with imatinib mesylate at dose of 1.25 mg/m2 twice a day for 14 days on 24 chronic phase CML patients in of. Complete blood and genetic response at cellular level of 66 and 55% respectively was achieved [191, 197]. Another study comprising of 225 patients with CML were evaluated for HHT at dose of 2.5 mg/m2 and ara-c combination treatment. Complete hematologic response of 81% was achieved upon this treatment [198, 199]. Combination treatment therapy using 2.5 mg/m2 of HHT with interferon alpha showed complete blood response at rate of 89% and cytogenetic response rate of 57% in CML patients in early chronic phase [200]. Average complete blood response rate of 94% and cellular genetic response rate of 74% were demonstrated in 90 CML patients receiving either HHT, interferon alpha and ara-C [201].
Omacetaxine mepesuccinate, in addition to being used in CML treatment, has also found its place in treatment of acute and multiple myeloma. Study conducted with continuous administration of HHT (5 mg/m2) for 9 days in 16 patients with myelodysplastic syndrome and 12 patients with myelodyplastic syndrome exhibited response rate of 28%. Complete and fractional remission in seven and one patient respectively was observed [202]. Phase II pilot study consisting of HHT at dose of 2.5 mg/m2 when given in form of infusion for 7 days followed by maintenance dose failed to show any response in eight patients [203].
Study was conducted in 66 patients with relapsed acute myeloid leukemia or blastic phase CML to evaluate efficacy of HTT. 16% of patients showed sign of complete remission. Two patients that were resistant to cytarbine showed complete remission whereas 11 patients resistant to cytarbine did not respond to treatment with HTT [204].
Clinical trials clearly justify the potential of HHT or its semi synthetic form in treatment of CML and other blood related disorders. However, along with its positive response hematological side effects like granulocytopenia, thrombocytopenia, myelosupression and non haemotological toxicities like diarrhea, fatigue, nausea, head ache chest pain etc. were commonly observed during course of treatment.
Gambogic acid
Gambogic acid (Fig. 2b) is a phytoconstituent belonging to class of xanthones which was isolated in form of gum resin form Garcinia hanburryi (also known as mangosteen). Xanthones, is a class of secondary metabolites isolated from plant, fungi and lichens, exhibits wide spectrum of activities such as anti-cancer, anti inflammatory, anti diabetic etc. [205,206,207,208]. Presently, the Chinese FDA has approved gambogic acid and phase II clinical trials are underway [209]. Development of imatinib mesylate resistance due to presence of BCR-ABL T3151 mutation in CML cells has led to search of novel therapeutic agent/s. Shi X group demonstrated the apoptotic effect of gambogic acid on CML cells, mononuclear cells from imatinib mesylate -sensitive or -resistant patients and in xenograft tumor model bearing T315I-BCR-ABL genes or wild-type BCR-ABL. It was observed that gambogic acid successfully induced apoptosis along with inhibition of cell proliferation in cell lines sensitive or resistant (bearing KBM5-T315I mutation as well as in the mononuclear cells from imatinib mesylate resistant patients) to imatinib mesylate. In xenograft model gambogic acid reduced tumor growth in nude mice harboring T315I-BCR-ABL mutation [209]. Structure-activity relationship (SAR) study by Sun et al., [210] showed that the caged 4-oxa-tricyclo[4.3.1.03,7]dec-2-one xanthone scaffold was the key pharmacophoric motif of gambogic acid. Molecular docking studies have shown that gambogic acid and its derivatives can bind to the ATP binding pocket of IKKβ and make several H-bonds with the hinge region of the enzyme, leading to inhibition of IKKβ. But there are no SAR studies with gambogic acid on any of the NRTKs.
Haishengsu
Haishengsu is a protein molecule with molecular weight of approximately 15KDa obtained from shell fish Tegillarca granosa. This compound is expected to help improve clinical results in the case of patients with renal and lung cancer when used alone or as an adjuvant with conventional chemotherapeutic drug/s [211, 212]. In in vivo study mice bearing drug resistant leukemia cell lineK562/ADM(Adriamycin) Haishengsu acted by repressing levels of multi drug resistant gene-1 (mdr1), BCR-ABL and sorcin at dose of 1800 mg/kg which was significant in comparison to group that received no drug (control) and that received adriamycin and haishengsu in combination [213].
Triptolide
Triptolide (Fig. 2c) is isolated from leaves of Tripterygium wilfordii was studied for its effect on KBM5 cell lines (wild-type BCR-ABL, T315I mutant BCR-ABL) and on peripheral blood mono nuclear cells from imatinib mesylate resistant patients. Triptolide induced time and dose dependent apoptosis in KBM5 cells and in peripheral blood mono nuclear cells. Induction of apoptosis was accompanied with decreased expression of BCR-ABL, phosphorylated STAT5, CrkL and Erk1/2. In vivo study, using imatinib mesylate -resistant CML cells in nude mouse xenograft model triptolide inhibited tumor proliferation without encouraging much appreciable changes in bodyweight. Immnuno histochemical analysis supported claim of triptolide in downregulating BCR-ABL [214]. Synthesis and biological activity studies of Triptolide derivatives have shown that the C-14β–OH group, C-9,11-β-epoxide group, C-12,13-α-epoxide group, C-7,8-β-epoxide group, the 5-membered lactone ring and the C-5,6-position are critical for the cytotoxicity and antitumor activities of Triptolide [215]. But there are no specific studies relating to NRTKs.
Curcumin
Curcumin (Fig. 2d) is an alkaloid isolated from various Curcuma species decreased viability of cells and promoted apoptosis in cells isolated from B6 mice expressing wild-type BCR-ABL (B6p210) and T315I mutant (B6T315I) human leukemic cells. B6p210 cells bearing p210 oncogene was found to be more susceptible to treatment then compared to T315I mutant. Western blot and transcription factor assay revealed occurrence of apoptosis via inhibition of c-abl and NF-kB along with its downstream targets. Beside this upregulation of p53 was also observed in both B6p210 and B6T315I cells. Improvement in survival rate and white blood cells/GFP positive cell counts compared to control was observed in mice with B-ALL [216]. Studies with synthetic derivatives shows the involvement of phenolic OH group in the antioxidant activity [217]; methoxy group in on inflammatory responses and NF-κB signaling [218] in the biological activities of curcumin, while there are no SAR studies relating its function to the NRTKs.
Berbamine
One of the major component isolated from chinese herbal medicine is Berberis amurensis. It is an effective calcium channel blocker [219]. Besides being used as calcium channel blocker it has promising effects against chronic myeloid leukemia, breast cancer, and melanoma [220,221,222]. Wei et al., demonstrated the ability of berbamine (Fig. 2e) to reverse mdr-1 along with reduced expression of P-glycoprotein both in in vitro and in vivo models [220]. Similar results were observed on use of 4-chlorobenzoyl berbamine (BBD9) an analogue of berbamine in imatinib mesylate resistant cells (K562/IR) in vitro and in vivo. BBD9 lowered expression of p210BCR-ABL, IKKα, nuclear NF-κB p65 along with its downstream target [223, 224].
Tetrandrine
Tetrandrine (Fig. 2f) belongs to class of bis-benxylisoquinoline alkaloid isolated from Stephania tetrandra. Although tetrandrine has gained importance due its ability to inhibit several tumor cells in vitro but poor solubility has limited its use. Xu-Xh et al., 2012 studied effect of salt form of tetrandrine (Tetrandrine citrate) on imatanib resistant K562 cells bearing high expression levels of p210(BCR-ABL). Decrease in expression levels of p210(BCR-ABL), β-catenin and BCR-ABL at mRNA level were observed in imatinib mesylate resistant K562 cells in vitro. Nude mice bearing imatinib mesylate resistant K562 cells showed complete sign of regression with no symptoms of toxicity when orally administered with tetrandrine citrate at dose of 100 mg/kg body weight [225]. SAR study unveiled the role of -OCH3 group present in a particular benzene ring of tetrandrine in the regulation of non-voltage-operated Ca2+ entry and release of intracellular Ca2+ in human promyelocytic leukemia cells [226], while there are no studies relating its structure to NRTKs.
Oroxylin A
Oroxylin (Fig. 2g) (5,7-dihydroxy-6-methoxyflavone) is an O-methylated flavonoid isolated from herb Scutellariae baicalensis when studied in combination with imatinib mesylate resulted in marked depletion of pY705-STAT3 along with its downstream targets p- glycoprotein in imatinib mesylate resistant K562 cells. NOD/SCID mice bearing K562 cells demonstrated decrease in tumor volume and weight significantly in oroxylin A (80 mg/kg) and imatinib mesylate (200 mg/kg) combined group compared to control and drug alone group [227].
Chlorgenic acid
Chlorgenic acid (Fig. 2h)is isolated from leaves of Piper betel promoted cell death by hindering expression of BCR-ABL and c-Abl kinases through activation of p38 and ERK-MAP kinase in cells bearing positive BCR-ABL and in BCR-ABL–positive leukemic cells isolated from CML patients in vitro. Sodium salt of chlorogenic acid due to its better solubility was found to be more sensitive compared to parent compound. Reduction in cancer advancement in nude mice bearing K562 xenograft was observed with salt form of acid [228].
Celastrol
Celastrol (Fig. 2i) is an active triterpenoid isolated from Tripterygium wilfordii which reduces active levels of phospho BCR-ABL and total BCR-ABL in CML cells bearing wild-type BCR-ABL and in T315I mutant (BCR-ABL resistant to imatinib mesylate) cells. In vivo study demonstrated effect of celastrol in reducing size and weight of tumor in imatinib mesylate resistant and imatinib mesylate sensitive cells nude xenograt model. Reduced levels of c-abl and BCR-ABL were further confirmed by immunohistochemical analysis. Combinational therapy using celastrol and 17-AAG (tanespimycin or geldanamycin) for 72 h showed synergistic/co-additive inhibitory effect [229].
Pristemerin
Pristemerin (Fig. 2j) is a quinonemethide triterpenoid isolated from Celestraceae and Hippocrateaceae family species promoted cell death by inhibiting growth of CML cells. Pristemerin induced dose dependent decrease in levels of p-BCR-ABL and total BCR-ABL at protein and mRNA levels as detected in imatinib mesylate sensitive (KBM5), imatinib mesylate resistant (KBM5-T3151) cells lines and in K562 cells. Concomitantly phosphorylation of CRKL, STAT5, AKT were also decreased with little or minimal effect on total AKT and STAT5. Significant tumor growth inhibition was observed compared to control in imatinib mesylate resistant BCR-ABL-T3151 xenografts in nude mice. Authors also confirmed that BCR-ABL reticence preceded apoptosis [230].
Herbimycin A
Antibiotic herbimycin A (Fig. 2k) is isolated from culture filtrate of Streptomyces species MH237-Cf-8, at their non cytotoxic concentration reduced levels of p210c-abl and induced erythroid differentiation in K562 and KU812 cells obtained from Ph + leukemic patients [231]. Similar results were observed in study conducted by same group of researcher wherein treatment with herbimycin A induced differentiation and prolonged survival time of nude mice inoculated with C1 cells expressing high level of protein tyrokinase [232, 233]. Herbimycin A and its synthetic analogue 17-cyclopropylamino-herbimycin A and 4,5-dichloro-herbimycin inactivated various tyrokinase like src, c-abl, BCR-ABL [234].
Woodfordin I
Woodfordin I (Fig. 2l), is a macrocyclic ellagi tannin dimer isolated from Wodordia fruticose and Denbinobin isolated from Cannabis sativa reduced expression of p120 c-Abl, p210 BCR-ABL, c-Abl and BCR-ABL in K562 human leukemic cells [235, 236].
Oridonin
Oridonin (Fig. 2m) is a diterpenoid isolated from Rabdosia rubescens inhibited lyn and abl levels along with downstream target Akt/mTOR, Raf/MEK/ERK and STAT5 in Ph + ALL cell line and primary specimens from Ph + ALL patients. Oridonin with imatinib mesylate exerted in tandem effects by overcoming imatinib mesylate problem of upregulating Akt/mTOR and lyn signaling [237].
Substantial evidences focusing on the potential of numerous phytoconstituents in inhibiting carcinogenis using in vitro and in vivo models in diverse cell systems have been published [238, 239]. Phytoconstituents like apigenin, resveratrol etc. that are found in wide range of fruits and vegetables and are gaining prominence owing to its ability to induce apoptosis via loss of mitochondrial membrane potential and caspase activation in K562 sensitive and K562/IMA3 (K562 cell resistant to 3uM imatinib mesylate) cells [240,241,242,243]. In addition to this and above mentioned natural compounds like fiestin, hesperidin, virosecurinine, cryptotanshinone, quercetin, genistein vincristine, and many more had competence when used alone or in combination with other tyrosine kinase inhibitor to down regulate BCR/ABL and lyn levels in cells developing resistance to imatinib mesylate and CML patients [244,245,246,247,248,249].
Conclusion and Future Perspectives
Non receptor tyrosine kinase are involved in multiple signaling pathways that regulate vital functions such as cell proliferation and differentiation, and plays a role in human neoplasms, inflammatory and autoimmune diseases. Clinical use of highly successful tyrosine kinase inhibitors (such as imatinib Mesylate, Herceptin and Gefitinib) endorse the potential of targeted cancer therapy using specific NRTK inhibitors. Targeted therapy has the advantages of being less toxic than traditional cytotoxic chemotherapy due to specificity for cancer cells. The best example is imatinib mesylate, a maximum limit of dose could not be identified during the phase 1 clinical trials. Unfortunately, even targeted therapy with the FDA approved small molecule NRTK inhibitor, omacetaxin mepesuccinate, have hematological side effects and other drawbacks. Some of these side effects may be due to the inhibition of other related tyrosine kinases present in normal cells and therefore extremely difficult to avoid completely. Nevertheless, additional knowledge of the side effects will make possible to develop better targeted drugs that are able to avoid these limitations.
In the case of general TK inhibitors like imatinib mesylate, development of resistance (due to point mutations or gene amplification) has become the major challenge. Similarly, resistance towards NRTK inhibitors could also develop in patients. A long term strategy to design rapid and efficient biochemical and cell based high throughput assay for screening of novel kinase inhibitors is needed. Implementation of bioinformatics based methodologies (structure based drug design, based on the current knowledge of the three dimensional structures of target kinases, mathematical quantitative modeling of cancer progression and drug response, etc) can hasten the process of screening several natural compounds through the drug discovery process.
Although several natural compounds have proven their efficacy in in vitro and in vivo models as potent tyrokinase inhibitors, in-detail research is still required to establish natural compounds as lead molecules for clinical trial. Till date only one single natural compound, Homoharringtonine, has been able to successfully finish clinical trials and receive an FDA approval. A key obstacle in the development of a specific inhibitor is the variation in efficacy observed in the cell line based experiments and rodent models during drug discovery phase leading to the final efficacy in patients. NRTK inhibitors may well add invaluable contribution to treatments in combination with conventional chemotherapy.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- ara-C:
-
Cytarabine
- CML:
-
Chronic myeloid leukemia
- CRIB:
-
Cdc42/Rac-interactive domain
- CSK:
-
C-terminal Src kinase
- EGFR:
-
Epidermal growth factor receptor
- FAK:
-
Focal adhesion kinase
- F-BAR:
-
FCH-Bin–Amphiphysin–Rvs domain
- F-BD:
-
Filamentous actin binding domain
- FCH:
-
Fes/Fer/Cdc-42-interacting protein homology
- HHT:
-
Homoharingtonine
- IFN:
-
Interferon
- JAK:
-
Janus kinase
- JMML:
-
Juvenile myelomonocytic leukemia
- NRTK:
-
Non receptor tyrosine kinase
- PDGFR:
-
Platelet-derived growth factor receptor
- Ph:
-
Philadelphia chromosome
- PH:
-
Pleckstrin homology
- RTK:
-
Receptor tyrosine kinase
- SFK:
-
Src family of tyrosine kinase
- STAT:
-
Signal transducer and activator of transcription
- STK:
-
Serine-threonine kinase
- TK:
-
Tyrosine Kinase
- TKI:
-
Tyrosine kinase inhibitor
References
Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–98.
Scheijen B, Griffin JD. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene. 2002;21:3314–33.
Chase A, Cross NC. Signal transduction therapy in haematological malignancies: identification and targeting of tyrosine kinases. Clin Sci (Lond). 2006;111:233–49.
Paul MK, Mukhopadhyay AK. Tyrosine kinase - Role and significance in Cancer. Int J Med Sci. 2004;1:101–15.
Kosior K, Lewandowska-Grygiel M, Giannopoulos K. Tyrosine kinase inhibitors in hematological malignancies. Postepy Hig Med Dosw (Online). 2011;65:819–28.
Wadleigh M, DeAngelo DJ, Griffin JD, Stone RM. After chronic myelogenous leukemia: tyrosine kinase inhibitors in other hematologic malignancies. Blood. 2005;105:22–30.
Rossi JF. Targeted Therapies in Adult B-Cell Malignancies. Biomed Res Int. 2015;2015:217593.
Al-Obeidi FA, Lam KS. Development of inhibitors for protein tyrosine kinases. Oncogene. 2000;19:5690–701.
Lahiry P, Torkamani A, Schork NJ, Hegele RA. Kinase mutations in human disease: interpreting genotype–phenotype relationships. Nat Rev Genet. 2010;11:60–74.
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2.
Pawson T. Regulation and targets of receptor tyrosine kinases. Eur J Cancer. 2002;38(Suppl 5):S3–10.
Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20.
Welch PJ, Wang JY. A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell. 1993;75:779–90.
Yin B. Focal adhesion kinase as a target in the treatment of hematological malignancies. Leuk Res. 2011;35:1416–8.
Elias D, Ditzel HJ. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacol Res. 2015;100:250–4.
Prieto-Echague V, Miller WT. Regulation of ack-family nonreceptor tyrosine kinases. J Signal Transduct. 2011;2011:742372.
Jacobs S, Hansen F, Kasl S, Ostfeld A, Berkman L, Kim K. Anxiety disorders during acute bereavement: risk and risk factors. J Clin Psychiatry. 1990;51:269–74.
Mano H. Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 1999;10:267–80.
Futterer K, Wong J, Grucza RA, Chan AC, Waksman G. Structural basis for Syk tyrosine kinase ubiquity in signal transduction pathways revealed by the crystal structure of its regulatory SH2 domains bound to a dually phosphorylated ITAM peptide. J Mol Biol. 1998;281:523–37.
Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402.
Geahlen RL. Syk and pTyr'd: Signaling through the B cell antigen receptor. Biochim Biophys Acta. 2009;1793:1115–27.
Schenk PW, Snaar-Jagalska BE. Signal perception and transduction: the role of protein kinases. Biochim Biophys Acta. 1999;1449:1–24.
Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–23.
Edelman AM, Blumenthal DK, Krebs EG. Protein serine/threonine kinases. Annu Rev Biochem. 1987;56:567–613.
Capra M, Nuciforo PG, Confalonieri S, Quarto M, Bianchi M, Nebuloni M, Boldorini R, Pallotti F, Viale G, Gishizky ML, et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res. 2006;66:8147–54.
Guicciardi ME, Gores GJ. AIP1: a new player in TNF signaling. J Clin Invest. 2003;111:1813–5.
Chalandon Y, Schwaller J. Targeting mutated protein tyrosine kinases and their signaling pathways in hematologic malignancies. Haematologica. 2005;90:949–68.
Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell. 2008;100:617–31.
Wang J, Pendergast AM. The emerging role of ABL kinases in solid tumors. Trends in cancer. 2015;1:110–23.
Antoku S, Saksela K, Rivera GM, Mayer BJ. Essential role of c-Abl PxxP motifs and its interactions with Crk and Nck adaptors in cell spreading. J Cell Sci. 2008;121:3071.
Hantschel O. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes & cancer. 2012;3:436–46.
Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. 2013;13:559–71.
Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal. 2010;3:re6.
Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci. 2016;129:9–16.
Pluk H, Dorey K, Superti-Furga G. Autoinhibition of c-Abl. Cell. 2002;108:247–59.
Chen S, O'Reilly LP, Smithgall TE, Engen JR. Tyrosine phosphorylation in the SH3 domain disrupts negative regulatory interactions within the c-Abl kinase core. J Mol Biol. 2008;383:414–23.
Cao C, Li Y, Leng Y, Li P, Ma Q, Kufe D. Ubiquitination and degradation of the Arg tyrosine kinase is regulated by oxidative stress. Oncogene. 2005;24:2433.
Meyn MA, Wilson MB, Abdi FA, Fahey N, Schiavone AP, Wu J, Hochrein JM, Engen JR, Smithgall TE. Src family kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J Biol Chem. 2006;281:30907–16.
Li S, Ilaria RL, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia–like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189:1399–412.
Advani AS, Pendergast AM. Bcr–Abl variants: biological and clinical aspects. Leuk Res. 2002;26:713–20.
Panjarian S, Iacob RE, Chen S, Engen JR, Smithgall TE. Structure and dynamic regulation of Abl kinases. J Biol Chem. 2013;288:5443–50.
Lamontanara AJ, Georgeon S, Tria G, Svergun DI, Hantschel O. The SH2 domain of Abl kinases regulates kinase autophosphorylation by controlling activation loop accessibility. Nat Commun. 2014;5:5470.
Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner K-U, Superti-Furga G, Sexl V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012;8:285–93.
Leoni V, Biondi A: Tyrosine kinase inhibitors in BCR-ABL positive acute lymphoblastic leukemia. Haematologica; 2015.
Gopal MM, Kotwal J, Kapoor R. Acute Myeloid Leukemia with BCR/ABL Fusion Chimera. Indian J Hematol Blood Transfusion. 2014;30:280–2.
Soupir CP, Vergilio J-A, Cin PD, Muzikansky A, Kantarjian H, Jones D, Hasserjian RP. Philadelphia chromosome–positive acute myeloid leukemia: a rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am J Clin Pathol. 2007;127:642–50.
Li J, Smithgall TE. Fibroblast transformation by Fps/Fes tyrosine kinases requires Ras, Rac, and Cdc42 and induces extracellular signal-regulated and c-Jun N-terminal kinase activation. J Biol Chem. 1998;273:13828–34.
Voisset E, Lopez S, Dubreuil P, De Sepulveda P. The tyrosine kinase FES is an essential effector of KIT D816V proliferation signal. Blood. 2007;110:2593–9.
Hellwig S, Miduturu CV, Kanda S, Zhang J, Filippakopoulos P, Salah E, Deng X, Choi HG, Zhou W, Hur W. Small-molecule inhibitors of the c-Fes protein-tyrosine kinase. Chem Biol. 2012;19:529–40.
Condorelli F, Stec-Martyna E, Zaborowska J, Felli L, Gemmi I, Ponassi M, Rosano C. Role of the non-receptor tyrosine kinase fes in cancer. Curr Med Chem. 2011;18:2913–20.
Sangrar W, Zirgnibl RA, Gao Y, Muller WJ, Jia Z, Greer PA. An identity crisis for fps/fes: oncogene or tumor suppressor? Cancer Res. 2005;65:3518–22.
Voisset E, Lopez S, Chaix A, Georges C, Hanssens K, Prebet T, Dubreuil P, De Sepulveda P. FES kinases are required for oncogenic FLT3 signaling. Leukemia. 2010;24:721.
Feldman RA, Lowy DR, Vass WC, Velu TJ. A highly efficient retroviral vector allows detection of the transforming activity of the human c-fps/fes proto-oncogene. J Virol. 1989;63:5469–74.
Furqan M, Mukhi N, Lee B, Liu D. Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomark Res. 2013;1:5.
Haan C, Is'harc H, Hermanns HM, Schmitz-Van De Leur H, Kerr IM, Heinrich PC, Grotzinger J, Behrmann I. Mapping of a region within the N terminus of Jak1 involved in cytokine receptor interaction. J Biol Chem. 2001;276:37451–8.
Wilks AF. The JAK kinases: not just another kinase drug discovery target. Semin Cell Dev Biol. 2008;19:319–28.
O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–28.
Levine RL, Gilliland DG. JAK-2 mutations and their relevance to myeloproliferative disease. Curr Opin Hematol. 2007;14:43–7.
Springuel L, Renauld JC, Knoops L. JAK kinase targeting in hematologic malignancies: a sinuous pathway from identification of genetic alterations towards clinical indications. Haematologica. 2015;100:1240–53.
Valentino L, Pierre J. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol. 2006;71:713–21.
Carter-Su C, Smit LS. Signaling via JAK tyrosine kinases: growth hormone receptor as a model system. Recent Prog Horm Res. 1998;53:61–82. discussion 82-63
Vainchenker W, Constantinescu S. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32:2601–13.
Vera J, Rateitschak K, Lange F, Kossow C, Wolkenhauer O, Jaster R. Systems biology of JAK-STAT signalling in human malignancies. Prog Biophys Mol Biol. 2011;106:426–34.
Constantinescu SN, Girardot M, Pecquet C. Mining for JAK-STAT mutations in cancer. Trends Biochem Sci. 2008;33:122–31.
Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ, Yoo NJ, Lee SH. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14:3716–21.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61.
Hornakova T, Chiaretti S, Lemaire MM, Foa R, Ben Abdelali R, Asnafi V, Tartaglia M, Renauld JC, Knoops L. ALL-associated JAK1 mutations confer hypersensitivity to the antiproliferative effect of type I interferon. Blood. 2010;115:3287–95.
Xiang Z, Zhao Y, Mitaksov V, Fremont DH, Kasai Y, Molitoris A, Ries RE, Miner TL, McLellan MD, DiPersio JF, et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood. 2008;111:4809–12.
Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, Ariola C, Fodale V, Clappier E, Paoloni F, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205:751–8.
Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106:9414–8.
Tefferi A. JAK and MPL mutations in myeloid malignancies. Leuk Lymphoma. 2008;49:388–97.
Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, Kon A, Sanada M, Chiba K, Tanaka H, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45:937–41.
Walters DK, Mercher T, Gu TL, O'Hare T, Tyner JW, Loriaux M, Goss VL, Lee KA, Eide CA, Wong MJ, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10:65–75.
Bouchekioua A, Scourzic L, De Wever O, Zhang Y, Cervera P, Aline-Fardin A, Mercher T, Gaulard P, Nyga R, Jeziorowska D. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia. 2014;28:338.
Bains T, Heinrich MC, Loriaux MM, Beadling C, Nelson D, Warrick A, Neff TL, Tyner JW, Dunlap J, Corless CL, Fan G. Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia. 2012;26:2144–6.
Sanda T, Tyner JW, Gutierrez A, Ngo VN, Glover J, Chang BH, Yost A, Ma W, Fleischman AG, Zhou W, et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013;3:564–77.
Joos S, Kupper M, Ohl S, von Bonin F, Mechtersheimer G, Bentz M, Marynen P, Moller P, Pfreundschuh M, Trumper L, Lichter P. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000;60:549–52.
Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–62.
Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105:13520–5.
Manser E, Leung T, Salihuddin H, Tan L, Lim L. A non-receptor tyrosine kinase that inhibits the GTPase activity of p21cdc42. Nature. 1993;363:364–7.
Mahajan K, Mahajan NP. ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers. Oncogene. 2015;34:4162–7.
Mahajan K, Mahajan NP. Shepherding AKT and androgen receptor by Ack1 tyrosine kinase. J Cell Physiol. 2010;224:327–33.
Grovdal LM, Johannessen LE, Rodland MS, Madshus IH, Stang E. Dysregulation of Ack1 inhibits down-regulation of the EGF receptor. Exp Cell Res. 2008;314:1292–300.
Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Rivera C, et al. Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS One. 2010;5:e9646.
Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005;65:10514–23.
Xie B, Zen Q, Wang X, He X, Xie Y, Zhang Z, Li H. ACK1 promotes hepatocellular carcinoma progression via downregulating WWOX and activating AKT signaling. Int J Oncol. 2015;46:2057–66.
Xu SH, Huang JZ, Chen M, Zeng M, Zou FY, Chen, Yan GR: Amplification of ACK1 promotes gastric tumorigenesis via ECD-dependent p53 ubiquitination degradation. Oncotarget 2017, 8:12705–12716.
Mahajan K, Mahajan NP. ACK1 tyrosine kinase: targeted inhibition to block cancer cell proliferation. Cancer Lett. 2013;338:185–92.
Liu Z, Liu Z, Zhang Y, Li Y, Liu B, Zhang K. miR-24 represses metastasis of human osteosarcoma cells by targeting Ack1 via AKT/MMPs pathway. Biochem Biophys Res Commun. 2017;486:211–7.
Lv C, Zhao X, Gu H, Huang L, Zhou S, Zhi F. Involvement of Activated Cdc42 Kinase1 in Colitis and Colorectal Neoplasms. Med Sci Monit. 2016;22:4794–802.
Xu SH, Huang JZ, Xu ML, Yu G, Yin XF, Chen D, Yan GR. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J Pathol. 2015;236:175–85.
Maxson JE, Abel ML, Wang J, Deng X, Reckel S, Luty SB, Sun H, Gorenstein J, Hughes SB, Bottomly D, et al. Identification and Characterization of Tyrosine Kinase Nonreceptor 2 Mutations in Leukemia through Integration of Kinase Inhibitor Screening and Genomic Analysis. Cancer Res. 2016;76:127–38.
Nonami A, Sattler M, Weisberg E, Liu Q, Zhang J, Patricelli MP, Christie AL, Saur AM, Kohl NE, Kung AL, et al. Identification of novel therapeutic targets in acute leukemias with NRAS mutations using a pharmacologic approach. Blood. 2015;125:3133–43.
Hoehn GT, Stokland T, Amin S, Ramirez M, Hawkins AL, Griffin CA, Small D, Civin CI. Tnk1: a novel intracellular tyrosine kinase gene isolated from human umbilical cord blood CD34+/Lin-/CD38- stem/progenitor cells. Oncogene. 1996;12:903–13.
Hoare K, Hoare S, Smith OM, Kalmaz G, Small D, Stratford May W. Kos1, a nonreceptor tyrosine kinase that suppresses Ras signaling. Oncogene. 2003;22:3562–77.
Azoitei N, Brey A, Busch T, Fulda S, Adler G, Seufferlein T. Thirty-eight-negative kinase 1 (TNK1) facilitates TNFalpha-induced apoptosis by blocking NF-kappaB activation. Oncogene. 2007;26:6536–45.
Henderson MC, Gonzales IM, Arora S, Choudhary A, Trent JM, Von Hoff DD, Mousses S, Azorsa DO. High-throughput RNAi screening identifies a role for TNK1 in growth and survival of pancreatic cancer cells. Mol Cancer Res. 2011;9:724–32.
Cho NL, Lin CI, Du J, Whang EE, Ito H, Moore FD Jr, Ruan DT. Global tyrosine kinome profiling of human thyroid tumors identifies Src as a promising target for invasive cancers. Biochem Biophys Res Commun. 2012;421:508–13.
Kobayashi T, Nakamura S, Taniguchi T, Yamamura H. Purification and characterization of a cytosolic protein-tyrosine kinase from porcine spleen. Eur J Biochem. 1990;188:535–40.
Taniguchi T, Kobayashi T, Kondo J, Takahashi K, Nakamura H, Suzuki J, Nagai K, Yamada T, Nakamura S, Yamamura H. Molecular cloning of a porcine gene syk that encodes a 72-kDa protein-tyrosine kinase showing high susceptibility to proteolysis. J Biol Chem. 1991;266:15790–6.
Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW 3rd, Su AI. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130.
Shin G, Kang TW, Yang S, Baek SJ, Jeong YS, Kim SY. GENT: gene expression database of normal and tumor tissues. Cancer Inform. 2011;10:149–57.
Feng G, Wang X. Role of spleen tyrosine kinase in the pathogenesis of chronic lymphocytic leukemia. Leuk Lymphoma. 2014;55:2699–705.
Liu D, Mamorska-Dyga A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol. 2017;10:145.
Efremov DG, Laurenti L. The Syk kinase as a therapeutic target in leukemia and lymphoma. Expert Opin Investig Drugs. 2011;20:623–36.
Krisenko MO, Geahlen RL. Calling in SYK: SYK's dual role as a tumor promoter and tumor suppressor in cancer. Biochim Biophys Acta. 1853;2015:254–63.
Qiu L, Wang F, Liu S, Chen XL. Current understanding of tyrosine kinase BMX in inflammation and its inhibitors. Burns Trauma. 2014;2:121–4.
Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, Christensson B, Berglof A, Vihinen M, Nore BF, Smith CI. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228:58–73.
Akinleye A, Furqan M, Adekunle O. Ibrutinib and indolent B-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2014;14:253–60.
Tojo A. Kinase inhibitors against hematological malignancies. Nihon Rinsho. 2014;72:1118–24.
Molina-Cerrillo J, Alonso-Gordoa T, Gajate P, Grande E. Brutońs tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat Rev. 2017;58:41–50.
Zhong Y, Dong S, Strattan E, Ren L, Butchar JP, Thornton K, Mishra A, Porcu P, Bradshaw JM, Bisconte A, et al. Targeting interleukin-2-inducible T-cell kinase (ITK) and resting lymphocyte kinase (RLK) using a novel covalent inhibitor PRN694. J Biol Chem. 2015;290:5960–78.
Xu WD, Su LC, Xie QB, Zhao Y, Liu Y. Interleukin-2-inducible T-cell kinase expression and relation to disease severity in systemic lupus erythematosus. Clin Chim Acta. 2016;463:11–7.
Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate NKT cell maturation, cytokine production, and survival. J Immunol. 2008;180:3007–18.
Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol. 2005;23:549–600.
Finkelstein LD, Shimizu Y, Schwartzberg PL. Tec kinases regulate TCR-mediated recruitment of signaling molecules and integrin-dependent cell adhesion. J Immunol. 2005;175:5923–30.
Readinger JA, Mueller KL, Venegas AM, Horai R, Schwartzberg PL. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev. 2009;228:93–114.
Takeba Y, Nagafuchi H, Takeno M, Kashiwakura J, Suzuki N. Txk, a member of nonreceptor tyrosine kinase of Tec family, acts as a Th1 cell-specific transcription factor and regulates IFN-gamma gene transcription. J Immunol. 2002;168:2365–70.
Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73.
Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. 1994;12:635–73.
Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521.
Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–93.
Block H, Zarbock A. The role of the tec kinase Bruton's tyrosine kinase (Btk) in leukocyte recruitment. Int Rev Immunol. 2012;31:104–18.
Gottar-Guillier M, Dodeller F, Huesken D, Iourgenko V, Mickanin C, Labow M, Gaveriaux S, Kinzel B, Mueller M, Alitalo K, et al. The tyrosine kinase BMX is an essential mediator of inflammatory arthritis in a kinase-independent manner. J Immunol. 2011;186:6014–23.
Yu L, Simonson OE, Mohamed AJ, Smith CI. NF-kappaB regulates the transcription of protein tyrosine kinase Tec. FEBS J. 2009;276:6714–24.
Mano H, Ishikawa F, Nishida J, Hirai H, Takaku F. A novel protein-tyrosine kinase, tec, is preferentially expressed in liver. Oncogene. 1990;5:1781–6.
Chen C, Wang G, Zhang ZM, Xu W, Li Q, Hu Q, Wang D, Li ZP, Yang ZX, Suo JY, et al. The expression of Tec and the level of its phosphorylation in primary hepatic carcinomas. Zhonghua Gan Zang Bing Za Zhi. 2007;15:910–3.
Vanova T, Konecna Z, Zbonakova Z, La Venuta G, Zoufalova K, Jelinkova S, Varecha M, Rotrekl V, Krejci P, Nickel W, et al. Tyrosine Kinase Expressed in Hepatocellular Carcinoma, TEC, Controls Pluripotency and Early Cell Fate Decisions of Human Pluripotent Stem Cells via Regulation of Fibroblast Growth Factor-2 Secretion. Stem Cells. 2017;35:2050–9.
Ozkal S, Paterson JC, Tedoldi S, Hansmann ML, Kargi A, Manek S, Mason DY, Marafioti T. Focal adhesion kinase (FAK) expression in normal and neoplastic lymphoid tissues. Pathol Res Pract. 2009;205:781–8.
Bryant PW, Zheng Q, Pumiglia KM. Focal adhesion kinase is a phospho-regulated repressor of Rac and proliferation in human endothelial cells. Biology open. 2012;1:723–30.
Carter BZ, Mak PY, Wang X, Yang H, Garcia-Manero G, Mak DH, Mu H, Ruvolo VR, Qiu Y, Coombes K, et al. Focal Adhesion Kinase as a Potential Target in AML and MDS. Mol Cancer Ther. 2017;16:1133–44.
Recher C, Ysebaert L, Beyne-Rauzy O, Mansat-De Mas V, Ruidavets JB, Cariven P, Demur C, Payrastre B, Laurent G, Racaud-Sultan C. Expression of focal adhesion kinase in acute myeloid leukemia is associated with enhanced blast migration, increased cellularity, and poor prognosis. Cancer Res. 2004;64:3191–7.
Aly RM, Ghazy HF. High expression of GTPase regulator associated with the focal adhesion kinase (GRAF) is a favorable prognostic factor in acute myeloid leukemia. Blood Cells Mol Dis. 2014;53:185–8.
Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–9.
Sen B, Johnson FM. Regulation of SRC family kinases in human cancers. J Signal Transduct. 2011;2011:865819.
Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–27.
Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–58.
Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–68.
Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23:7969–78.
Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602.
Reynolds AB, Roczniak-Ferguson A. Emerging roles for p120-catenin in cell adhesion and cancer. Oncogene. 2004;23:7947–56.
Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004;23:8017–23.
Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–46.
Bourrie B, Brassard DL, Cosnier-Pucheu S, Zilberstein A, Yu K, Levit M, Morrison JG, Perreaut P, Jegham S, Hilairet S, et al. SAR103168: a tyrosine kinase inhibitor with therapeutic potential in myeloid leukemias. Leuk Lymphoma. 2013;54:1488–99.
Bunda S, Kang MW, Sybingco SS, Weng J, Favre H, Shin DH, Irwin MS, Loh ML, Ohh M. Inhibition of SRC corrects GM-CSF hypersensitivity that underlies juvenile myelomonocytic leukemia. Cancer Res. 2013;73:2540–50.
Miranda MB, Johnson DE. Signal transduction pathways that contribute to myeloid differentiation. Leukemia. 2007;21:1363–77.
Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000.
Gauld SB, Cambier JC. Src-family kinases in B-cell development and signaling. Oncogene. 2004;23:8001–6.
Warmuth M, Damoiseaux R, Liu Y, Fabbro D, Gray N. SRC family kinases: potential targets for the treatment of human cancer and leukemia. Curr Pharm Des. 2003;9:2043–59.
Ku M, Wall M, MacKinnon RN, Walkley CR, Purton LE, Tam C, Izon D, Campbell L, Cheng HC, Nandurkar H. Src family kinases and their role in hematological malignancies. Leuk Lymphoma. 2015;56:577–86.
Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012;8:1385–97.
Chan PC, Chen HC. p120RasGAP-mediated activation of c-Src is critical for oncogenic Ras to induce tumor invasion. Cancer Res. 2012;72:2405–15.
Congleton J, MacDonald R, Yen A. Src inhibitors, PP2 and dasatinib, increase retinoic acid-induced association of Lyn and c-Raf (S259) and enhance MAPK-dependent differentiation of myeloid leukemia cells. Leukemia. 2012;26:1180–8.
Zhang S, Yu D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol Sci. 2012;33:122–8.
Mayer EL, Krop IE. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin Cancer Res. 2010;16:3526–32.
Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–8.
Pene-Dumitrescu T, Smithgall TE. Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner. J Biol Chem. 2010;285:21446–57.
Malek RL, Irby RB, Guo QM, Lee K, Wong S, He M, Tsai J, Bryan F, Liu ET, Quackenbush J. Identification of Src transformation fingerprint in human colon cancer. Oncogene. 2002;21:7256.
Kato J, Takeya T, Grandori C, Iba H, Levy J, Hanafusa H. Amino acid substitutions sufficient to convert the nontransforming p60c-src protein to a transforming protein. Mol Cell Biol. 1986;6:4155–60.
Levy JB, Iba H, Hanafusa H. Activation of the transforming potential of p60c-src by a single amino acid change. Proc Natl Acad Sci. 1986;83:4228–32.
Saito YD, Jensen AR, Salgia R, Posadas EM. Fyn: a novel molecular target in cancer. Cancer. 2010;116:1629–37.
Singh MM, Howard A, Irwin ME, Gao Y, Lu X, Multani A, Chandra J. Expression and activity of Fyn mediate proliferation and blastic features of chronic myelogenous leukemia. PLoS One. 2012;7:e51611.
Gao Y, Howard A, Ban K, Chandra J. Oxidative stress promotes transcriptional up-regulation of Fyn in BCR-ABL1-expressing cells. J Biol Chem. 2009;284:7114–25.
Contri A, Brunati AM, Trentin L, Cabrelle A, Miorin M, Cesaro L, Pinna LA, Zambello R, Semenzato G, Donella-Deana A. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J Clin Invest. 2005;115:369–78.
Hussein K, von Neuhoff N, Busche G, Buhr T, Kreipe H, Bock O. Opposite expression pattern of Src kinase Lyn in acute and chronic haematological malignancies. Ann Hematol. 2009;88:1059–67.
Tibaldi E, Pagano MA, Frezzato F, Trimarco V, Facco M, Zagotto G, Ribaudo G, Pavan V, Bordin L, Visentin A, et al. Targeted activation of the SHP-1/PP2A signaling axis elicits apoptosis of chronic lymphocytic leukemia cells. Haematologica. 2017;102:1401–12.
Gattazzo C, Martini V, Frezzato F, Trimarco V, Tibaldi E, Castelli M, Facco M, Zonta F, Brunati AM, Zambello R, et al. Cortactin, another player in the Lyn signaling pathway, is over-expressed and alternatively spliced in leukemic cells from patients with B-cell chronic lymphocytic leukemia. Haematologica. 2014;99:1069–77.
ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia--focus on the B-cell receptor. Clin Cancer Res. 2014;20:548–56.
Zonta F, Pagano MA, Trentin L, Tibaldi E, Frezzato F, Trimarco V, Facco M, Zagotto G, Pavan V, Ribaudo G, et al. Lyn sustains oncogenic signaling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood. 2015;125:3747–55.
Advani G, Lim YC, Catimel B, Lio DSS, Ng NL, Chüeh AC, Tran M, Anasir MI, Verkade H, Zhu H-J. Csk-homologous kinase (Chk) is an efficient inhibitor of Src-family kinases but a poor catalyst of phosphorylation of their C-terminal regulatory tyrosine. Cell Communication and Signaling. 2017;15:29.
Bodensteiner DC, Doolittle GC. Adverse haematological complications of anticancer drugs. Clinical presentation, management and avoidance. Drug Saf. 1993;8:213–24.
Sampi K, Taguchi T. Gastrointestinal toxicity induced by anticancer drugs--including new antiemetic drugs. Gan To Kagaku Ryoho. 1990;17:922–8.
Duwe G, Knitter S, Pesthy S, Beierle AS, Bahra M, Schmelzle M, Schmuck RB, Lohneis P, Raschzok N, Ollinger R, et al. Hepatotoxicity following systemic therapy for colorectal liver metastases and the impact of chemotherapy-associated liver injury on outcomes after curative liver resection. Eur J Surg Oncol. 2017;43:1668–81.
Lameire N. Nephrotoxicity of recent anti-cancer agents. Clin Kidney J. 2014;7:11–22.
Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–9.
Illmer T, Schaich M, Platzbecker U, Freiberg-Richter J, Oelschlagel U, von Bonin M, Pursche S, Bergemann T, Ehninger G, Schleyer E. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia. 2004;18:401–8.
Deenik W, van der Holt B, Janssen JJ, Chu IW, Valk PJ, Ossenkoppele GJ, van der Heiden IP, Sonneveld P, van Schaik RH, Cornelissen JJ. Polymorphisms in the multidrug resistance gene MDR1 (ABCB1) predict for molecular resistance in patients with newly diagnosed chronic myeloid leukemia receiving high-dose imatinib. Blood. 2010;116:6144–5. author reply 6145-6146
Deremer DL, Katsanevas K, Ustun C. Critical appraisal of nilotinib in frontline treatment of chronic myeloid leukemia. Cancer Manag Res. 2011;3:65–78.
Bastias G, Pantoja T, Leisewitz T, Zarate V. Health care reform in Chile. CMAJ. 2008;179:1289–92.
Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, Mestan J, Trappe J, Wartmann M, Fabbro D. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 1804;2010:445–53.
Ray A, Cowan-Jacob SW, Manley PW, Mestan J, Griffin JD. Identification of BCR-ABL point mutations conferring resistance to the Abl kinase inhibitor AMN107 (nilotinib) by a random mutagenesis study. Blood. 2007;109:5011–5.
Kinch MS, Haynesworth A, Kinch SL, Hoyer D. An overview of FDA-approved new molecular entities: 1827-2013. Drug Discov Today. 2014;19:1033–9.
Demain AL. Importance of microbial natural products and the need to revitalize their discovery. J Ind Microbiol Biotechnol. 2014;41:185–201.
Chen Y, Li S. Omacetaxine mepesuccinate in the treatment of intractable chronic myeloid leukemia. Onco Targets Ther. 2014;7:177–86.
Rosshandler Y, Shen AQ, Cortes J, Khoury HJ. Omacetaxine Mepesuccinate for Chronic Myeloid Leukemia. Expert Rev Hematol. 2016;9:419–24.
Chen Y, Hu Y, Michaels S, Segal D, Brown D, Li S. Inhibitory effects of omacetaxine on leukemic stem cells and BCR-ABL-induced chronic myeloid leukemia and acute lymphoblastic leukemia in mice. Leukemia. 2009;23:1446–54.
Tong H, Ren Y, Zhang F, Jin J. Homoharringtonine affects the JAK2-STAT5 signal pathway through alteration of protein tyrosine kinase phosphorylation in acute myeloid leukemia cells. Eur J Haematol. 2008;81:259–66.
Coude MM, Luycx O, Cariou ME, Maarek O, Dombret H, Cayuela JM, Rea D. Undetectable molecular residual disease after omacetaxine and nilotinib combination therapy in an imatinib-resistant chronic myeloid leukaemia patient harbouring the BCR-ABL1 T315I gatekeeper mutation. Br J Haematol. 2012;157:407–10.
Lü S, Wang J. Homoharringtonine and omacetaxine for myeloid hematological malignancies. J Hematol Oncol. 2014;7:2.
Cortes J, Digumarti R, Parikh PM, Wetzler M, Lipton JH, Hochhaus A, Craig AR, Benichou AC, Nicolini FE, Kantarjian HM, Omacetaxine 203 Study G. Phase 2 study of subcutaneous omacetaxine mepesuccinate for chronic-phase chronic myeloid leukemia patients resistant to or intolerant of tyrosine kinase inhibitors. Am J Hematol. 2013;88:350–4.
Marin D, Kaeda JS, Andreasson C, Saunders SM, Bua M, Olavarria E, Goldman JM, Apperley JF. Phase I/II trial of adding semisynthetic homoharringtonine in chronic myeloid leukemia patients who have achieved partial or complete cytogenetic response on imatinib. Cancer. 2005;103:1850–5.
Quintas-Cardama A, Kantarjian H, Garcia-Manero G, O'Brien S, Faderl S, Estrov Z, Giles F, Murgo A, Ladie N, Verstovsek S, Cortes J. Phase I/II study of subcutaneous homoharringtonine in patients with chronic myeloid leukemia who have failed prior therapy. Cancer. 2007;109:248–55.
Cortes J, Lipton JH, Rea D, Digumarti R, Chuah C, Nanda N, Benichou AC, Craig AR, Michallet M, Nicolini FE, et al. Phase 2 study of subcutaneous omacetaxine mepesuccinate after TKI failure in patients with chronic-phase CML with T315I mutation. Blood. 2012;120:2573–80.
O'Brien S, Kantarjian H, Koller C, Feldman E, Beran M, Andreeff M, Giralt S, Cheson B, Keating M, Freireich E, et al. Sequential homoharringtonine and interferon-alpha in the treatment of early chronic phase chronic myelogenous leukemia. Blood. 1999;93:4149–53.
Nicolini FE, Khoury HJ, Akard L, Rea D, Kantarjian H, Baccarani M, Leonoudakis J, Craig A, Benichou AC, Cortes J. Omacetaxine mepesuccinate for patients with accelerated phase chronic myeloid leukemia with resistance or intolerance to two or more tyrosine kinase inhibitors. Haematologica. 2013;98:e78–9.
Nicolini FE, Chomel JC, Roy L, Legros L, Chabane K, Ducastelle S, Nicolas-Virelizier E, Michallet M, Tigaud I, Magaud JP, et al. The durable clearance of the T315I BCR-ABL mutated clone in chronic phase chronic myelogenous leukemia patients on omacetaxine allows tyrosine kinase inhibitor rechallenge. Clin Lymphoma Myeloma Leuk. 2010;10:394–9.
Li YF, Liu X, Liu DS, Din BH, Zhu JB. The effect of homoharringtonine in patients with chronic myeloid leukemia who have failed or responded suboptimally to imatinib therapy. Leuk Lymphoma. 2009;50:1889–91.
Kantarjian HM, Talpaz M, Smith TL, Cortes J, Giles FJ, Rios MB, Mallard S, Gajewski J, Murgo A, Cheson B, O'Brien S. Homoharringtonine and low-dose cytarabine in the management of late chronic-phase chronic myelogenous leukemia. J Clin Oncol. 2000;18:3513–21.
Stone RM, Donohue KA, Stock W, Hars V, Linker CA, Shea T, Deangelo DJ, Marcucci G, Bloomfield CD, Larson RA, et al. A phase II study of continuous infusion homoharringtonine and cytarabine in newly diagnosed patients with chronic myeloid leukemia: CALGB study 19804. Cancer Chemother Pharmacol. 2009;63:859–64.
O'Brien S, Talpaz M, Cortes J, Shan J, Giles FJ, Faderl S, Thomas D, Garcia-Manero G, Mallard S, Beth M, et al. Simultaneous homoharringtonine and interferon-alpha in the treatment of patients with chronic-phase chronic myelogenous leukemia. Cancer. 2002;94:2024–32.
O'Brien S, Giles F, Talpaz M, Cortes J, Rios MB, Shan J, Thomas D, Andreeff M, Kornblau S, Faderl S, et al. Results of triple therapy with interferon-alpha, cytarabine, and homoharringtonine, and the impact of adding imatinib to the treatment sequence in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in early chronic phase. Cancer. 2003;98:888–93.
Feldman EJ, Seiter KP, Ahmed T, Baskind P, Arlin ZA. Homoharringtonine in patients with myelodysplastic syndrome (MDS) and MDS evolving to acute myeloid leukemia. Leukemia. 1996;10:40–2.
Daver N, Vega-Ruiz A, Kantarjian HM, Estrov Z, Ferrajoli A, Kornblau S, Verstovsek S, Garcia-Manero G, Cortes JE. A phase II open-label study of the intravenous administration of homoharringtonine in the treatment of myelodysplastic syndrome. Eur J Cancer Care (Engl). 2013;22:605–11.
Feldman E, Arlin Z, Ahmed T, Mittelman A, Puccio C, Chun H, Cook P, Baskind P. Homoharringtonine in combination with cytarabine for patients with acute myelogenous leukemia. Leukemia. 1992;6:1189–91.
Zhu X, Zhang H, Lin Y, Chen P, Min J, Wang Z, Xiao W, Chen B. Mechanisms of gambogic acid-induced apoptosis in non-small cell lung cancer cells in relation to transferrin receptors. J Chemother. 2009;21:666–72.
Mu R, Lu N, Wang J, Yin Y, Ding Y, Zhang X, Gui H, Sun Q, Duan H, Zhang L, et al. An oxidative analogue of gambogic acid-induced apoptosis of human hepatocellular carcinoma cell line HepG2 is involved in its anticancer activity in vitro. Eur J Cancer Prev. 2010;19:61–7.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19.
Zhao B, Shen H, Zhang L, Shen Y. Gambogic acid activates AMP-activated protein kinase in mammalian cells. Biochem Biophys Res Commun. 2012;424:100–4.
Shi X, Chen X, Li X, Lan X, Zhao C, Liu S, Huang H, Liu N, Liao S, Song W, et al. Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin Cancer Res. 2014;20:151–63.
Sun H, Chen F, Wang X, Liu Z, Yang Q, Zhang X, Zhu J, Qiang L, Guo Q, You Q. Studies on gambogic acid (IV): Exploring structure–activity relationship with IκB kinase-beta (IKKβ). Eur J Med Chem. 2012;51:110–23.
Li GY, Yu XM, Zhang HW, Zhang B, Wang CB, Xin YC, Yang CZ, Zhou RX, Wang LX. Haishengsu as an adjunct therapy to conventional chemotherapy in patients with non-small cell lung cancer: a pilot randomized and placebo-controlled clinical trial. Complement Ther Med. 2009;17:51–5.
Liu JZ, Chen SG, Zhang B, Wang CB, Zhao XW, Li GY, Wang LX. Effect of haishengsu as an adjunct therapy for patients with advanced renal cell cancer: a randomized and placebo-controlled clinical trial. J Altern Complement Med. 2009;15:1127–30.
Li GY, Liu JZ, Zhang B, Yang M, Chen SG, Hou M, Wang LX. Tegillarca granosa extract Haishengsu (HSS) suppresses expression of mdr1, BCR/ABL and sorcin in drug-resistant K562/ADM tumors in mice. Adv Med Sci. 2013;58:112–7.
Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q, Lu Z, Chen Q, Lai Y, Pan J. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res. 2009;15:1686–97.
Zhou Z-L, Yang Y-X, Ding J, Li Y-C, Miao Z-H. Triptolide: structural modifications, structure–activity relationships, bioactivities, clinical development and mechanisms. Nat Prod Rep. 2012;29:457–75.
William BM, Goodrich A, Peng C, Li S. Curcumin inhibits proliferation and induces apoptosis of leukemic cells expressing wild-type or T315I-BCR-ABL and prolongs survival of mice with acute lymphoblastic leukemia. Hematology. 2008;13:333–43.
Priyadarsini KI, Maity DK, Naik G, Kumar MS, Unnikrishnan M, Satav J, Mohan H. Role of phenolic OH and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic Biol Med. 2003;35:475–84.
Yang H, Du Z, Wang W, Song M, Sanidad KZ, Sukamtoh E, Zheng J, Tian L, Xiao H, Liu Z. Structure and Activity Relationship of Curcumin: Role of Methoxy Group in Anti-inflammatory and Anti-colitis Effects of Curcumin. J Agric Food Chem. 2017;65:4509–15.
Chai S, To KK, Lin G. Circumvention of multi-drug resistance of cancer cells by Chinese herbal medicines. Chin Med. 2010;5:26.
Wei YL, Xu L, Liang Y, Xu XH, Zhao XY. Berbamine exhibits potent antitumor effects on imatinib-resistant CML cells in vitro and in vivo. Acta Pharmacol Sin. 2009;30:451–7.
Karnam KC, Ellutla M, Bodduluru LN, Kasala ER, Uppulapu SK, Kalyankumarraju M, Lahkar M. Preventive effect of berberine against DMBA-induced breast cancer in female Sprague Dawley rats. Biomed Pharmacother. 2017;92:207–14.
Nam S, Xie J, Perkins A, Ma Y, Yang F, Wu J, Wang Y, Xu RZ, Huang W, Horne DA, Jove R. Novel synthetic derivatives of the natural product berbamine inhibit Jak2/Stat3 signaling and induce apoptosis of human melanoma cells. Mol Oncol. 2012;6:484–93.
Zhang YF, Xu GB, Gan YC, Xu XH, Xu RZ. Inhibitory effect of 4-chlorobenzoyl berbamine on imatinib-resistant K562 cells in vitro and in vivo. Nan Fang Yi Ke Da Xue Xue Bao. 2011;31:1997–2001.
Wei YL, Liang Y, Xu L, Zhao XY. The antiproliferation effect of berbamine on k562 resistant cells by inhibiting NF-kappaB pathway. Anat Rec (Hoboken). 2009;292:945–50.
Xu XH, Gan YC, Xu GB, Chen T, Zhou H, Tang JF, Gu Y, Xu F, Xie YY, Zhao XY, Xu RZ. Tetrandrine citrate eliminates imatinib-resistant chronic myeloid leukemia cells in vitro and in vivo by inhibiting Bcr-Abl/beta-catenin axis. J Zhejiang Univ Sci B. 2012;13:867–74.
Leung YM, Berdik M, Kwan CY, Loh TT. Effects of tetrandrine and closely related bis-benzylisoquinoline derivatives on cytosolic Ca2+ in human leukaemic HL-60 cells: a structure-activity relationship study. Clin Exp Pharmacol Physiol. 1996;23:653–9.
Li X, Miao H, Zhang Y, Li W, Li Z, Zhou Y, Zhao L, Guo Q. Bone marrow microenvironment confers imatinib resistance to chronic myelogenous leukemia and oroxylin A reverses the resistance by suppressing Stat3 pathway. Arch Toxicol. 2015;89:121–36.
Bandyopadhyay G, Biswas T, Roy KC, Mandal S, Mandal C, Pal BC, Bhattacharya S, Rakshit S, Bhattacharya DK, Chaudhuri U, et al. Chlorogenic acid inhibits Bcr-Abl tyrosine kinase and triggers p38 mitogen-activated protein kinase-dependent apoptosis in chronic myelogenous leukemic cells. Blood. 2004;104:2514–22.
Lu Z, Jin Y, Qiu L, Lai Y, Pan J. Celastrol, a novel HSP90 inhibitor, depletes Bcr-Abl and induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation. Cancer Lett. 2010;290:182–91.
Lu Z, Jin Y, Chen C, Li J, Cao Q, Pan J. Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-kappaB signaling and depleting Bcr-Abl. Mol Cancer. 2010;9:112.
Honma Y, Okabe-Kado J, Hozumi M, Uehara Y, Mizuno S. Induction of erythroid differentiation of K562 human leukemic cells by herbimycin A, an inhibitor of tyrosine kinase activity. Cancer Res. 1989;49:331–4.
Honma Y, Okabe-Kado J, Kasukabe T, Hozumi M, Kodama H, Kajigaya S, Suda T, Miura Y. Herbimycin A, an inhibitor of tyrosine kinase, prolongs survival of mice inoculated with myeloid leukemia C1 cells with high expression of v-abl tyrosine kinase. Cancer Res. 1992;52:4017–20.
Honma Y, Okabe-Kado J, Kasukabe T, Hozumi M, Kajigaya S, Suda T, Miura Y. Induction by some protein kinase inhibitors of differentiation of a mouse megakaryoblastic cell line established by coinfection with Abelson murine leukemia virus and recombinant SV40 retrovirus. Cancer Res. 1991;51:4649–55.
Sato S, Honma Y, Hozumi M, Hayashi Y, Matsuo Y, Shibata K, Omura S, Hino K, Tomoyasu S, Tsuruoka N. Effects of herbimycin A and its derivatives on growth and differentiation of Ph1-positive acute lymphoid leukemia cell lines. Leuk Res. 1994;18:221–8.
Liu MJ, Wang Z, Li HX, Wu RC, Liu YZ, Wu QY. Mitochondrial dysfunction as an early event in the process of apoptosis induced by woodfordin I in human leukemia K562 cells. Toxicol Appl Pharmacol. 2004;194:141–55.
Huang YC, Guh JH, Teng CM. Denbinobin-mediated anticancer effect in human K562 leukemia cells: role in tubulin polymerization and Bcr-Abl activity. J Biomed Sci. 2005;12:113–21.
Guo Y, Shan Q, Gong Y, Lin J, Yang X, Zhou R. Oridonin in combination with imatinib exerts synergetic anti-leukemia effect in Ph+ acute lymphoblastic leukemia cells in vitro by inhibiting activation of LYN/mTOR signaling pathway. Cancer Biol Ther. 2012;13:1244–54.
Kamei H, Koide T, Kojimam T, Hasegawa M, Terabe K, Umeda T, Hashimoto Y. Flavonoid-mediated tumor growth suppression demonstrated by in vivo study. Cancer Biother Radiopharm. 1996;11:193–6.
Caltagirone S, Rossi C, Poggi A, Ranelletti FO, Natali PG, Brunetti M, Aiello FB, Piantelli M. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int J Cancer. 2000;87:595–600.
Can G, Cakir Z, Kartal M, Gunduz U, Baran Y. Apoptotic effects of resveratrol, a grape polyphenol, on imatinib-sensitive and resistant K562 chronic myeloid leukemia cells. Anticancer Res. 2012;32:2673–8.
Wu X-p, Xiong M, Xu C-S, Duan L-N, Dong Y-Q, Luo Y, Niu T-H, Lu C-R. Resveratrol induces apoptosis of human chronic myelogenous leukemia cells in vitro through p38 and JNK-regulated H2AX phosphorylation. Acta Pharmacol Sin. 2015;36:353–61.
Solmaz S, Adan Gokbulut A, Cincin B, Ozdogu H, Boga C, Cakmakoglu B, Kozanoglu I, Baran Y. Therapeutic potential of apigenin, a plant flavonoid, for imatinib-sensitive and resistant chronic myeloid leukemia cells. Nutr Cancer. 2014;66:599–612.
Angulo P, Kaushik G, Subramaniam D, Dandawate P, Neville K, Chastain K, Anant S. Natural compounds targeting major cell signaling pathways: a novel paradigm for osteosarcoma therapy. J Hematol Oncol. 2017;10:10.
Adan A, Baran Y. Fisetin and hesperetin induced apoptosis and cell cycle arrest in chronic myeloid leukemia cells accompanied by modulation of cellular signaling. Tumour Biol. 2016;37:5781–95.
Zhang G, Li M, Han S, Chen D, Wang Y, Ye W, Ji Z. Induction of human chronic myeloid leukemia K562 cell apoptosis by virosecurinine and its molecular mechanism. Mol Med Rep. 2014;10:2365–71.
Ambrosino DM, Siber GR, Chilmonczyk BA, Jernberg JB, Finberg RW. An immunodeficiency characterized by impaired antibody responses to polysaccharides. N Engl J Med. 1987;316:790–3.
Deora AB, Miranda MB, Rao SG. Down-modulation of P210bcr/abl induces apoptosis/differentiation in K562 leukemic blast cells. Tumori. 1997;83:756–61.
Scappini B, Onida F, Kantarjian HM, Dong L, Verstovsek S, Keating MJ, Beran M. In vitro effects of STI 571-containing drug combinations on the growth of Philadelphia-positive chronic myelogenous leukemia cells. Cancer. 2002;94:2653–62.
Kano Y, Akutsu M, Tsunoda S, Mano H, Sato Y, Honma Y, Furukawa Y. In vitro cytotoxic effects of a tyrosine kinase inhibitor STI571 in combination with commonly used antileukemic agents. Blood. 2001;97:1999–2007.
Acknowledgements
The authors would like to thank the editors and anonymous reviewers.
Funding
The study was supported by grant from the Medical Research Center (to SU) (Grant # 16354/16), Hamad Medical Corporation, Doha, State of Qatar.
Availability of data and materials
Please contact author for data requests.
Author information
Authors and Affiliations
Contributions
KSS, KSP, IWA, SK, AQK, and MM performed literature review, compiled the data and write the manuscript. SD and SU made extensive contributions to the design of the manuscript and revised it critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Siveen, K.S., Prabhu, K.S., Achkar, I.W. et al. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and its Targeting by Natural Products. Mol Cancer 17, 31 (2018). https://doi.org/10.1186/s12943-018-0788-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0788-y