
Abstract
Background
Receptor protein tyrosine phosphatase beta/zeta (RPTPβ/ζ) is a chondroitin sulphate (CS) transmembrane protein tyrosine phosphatase and is a receptor for pleiotrophin (PTN). RPTPβ/ζ interacts with ανβ3 on the cell surface and upon binding of PTN leads to c-Src dephosphorylation at Tyr530, β3 Tyr773 phosphorylation, cell surface nucleolin (NCL) localization and stimulation of cell migration. c-Src-mediated β3 Tyr773 phosphorylation is also observed after vascular endothelial growth factor 165 (VEGF165) stimulation of endothelial cells and is essential for VEGF receptor type 2 (VEGFR2) - ανβ3 integrin association and subsequent signaling. In the present work, we studied whether RPTPβ/ζ mediates angiogenic actions of VEGF.
Methods
Human umbilical vein endothelial, human glioma U87MG and stably transfected Chinese hamster ovary cells expressing different β3 subunits were used. Protein-protein interactions were studied by a combination of immunoprecipitation/Western blot, immunofluorescence and proximity ligation assays, properly quantified as needed. RPTPβ/ζ expression was down-regulated using small interference RNA technology. Migration assays were performed in 24-well microchemotaxis chambers, using uncoated polycarbonate membranes with 8 μm pores.
Results
RPTPβ/ζ mediates VEGF165-induced c-Src-dependent β3 Tyr773 phosphorylation, which is required for VEGFR2-ανβ3 interaction and the downstream activation of phosphatidylinositol 3-kinase (PI3K) and cell surface NCL localization. RPTPβ/ζ directly interacts with VEGF165, and this interaction is not affected by bevacizumab, while it is interrupted by both CS-E and PTN. Down-regulation of RPTPβ/ζ by siRNA or administration of exogenous CS-E abolishes VEGF165-induced endothelial cell migration, while PTN inhibits the migratory effect of VEGF165 to the levels of its own effect.
Conclusions
These data identify RPTPβ/ζ as a cell membrane binding partner for VEGF that regulates angiogenic functions of endothelial cells and suggest that it warrants further validation as a potential target for development of additive or alternative anti-VEGF therapies.
Similar content being viewed by others


Background
Vascular endothelial growth factor A (VEGF) is a growth factor that activates several functions of endothelial cells, thus triggering angiogenesis and vascular permeability. It exists as different isoforms of 121, 145, 165, 189 and 206 amino acids, among which VEGF165 is dominant in terms of amount and biological activity. VEGF165 is over-expressed in a variety of human tumors, and its over-expression is correlated with progression, invasion, and metastasis of tumors. VEGF165 cell signaling leading to increased endothelial cell migration and tubular formation is mediated via vascular endothelial growth factor receptor 2 (VEGFR2), which interacts with co-receptors, such as ανβ3 integrin [1]. It has been shown that c-Src-mediated phosphorylation of β3 cytoplasmic tail tyrosine residues occurs in response to VEGF165 and is essential for VEGFR2-β3 integrin association and subsequent signaling [2,3]. Up to date, it remains unclear how c-Src is activated by VEGF165. The first step in c-Src activation requires dephosphorylation of its carboxy-terminus Tyr530 [4], suggesting that a tyrosine phosphatase may be involved.
Receptor protein tyrosine phosphatase beta/zeta (RPTPβ/ζ) is a member of the family of receptor-type transmembrane protein tyrosine phosphatases that interacts with several cell adhesion molecules, such as neutral cell adhesion molecule, neuron-glia cell adhesion molecule, tenascin and contactin. It also acts as a receptor for the soluble, heparin-binding growth factors midkine and pleiotrophin (PTN) [5]. RPTPβ/ζ and PTN are expressed in endothelial cells [6] and over-expressed in several types of cancer [5,7]. PTN binding to RPTPβ/ζ on endothelial cells leads to Tyr530 dephosphorylation and activation of c-Src, β3 Tyr773 phosphorylation, and PTN-induced endothelial cell migration and tube formation on matrigel [5,8]. Besides PTN, RPTPβ/ζ has been shown to be the putative receptor for the vacuolating cytotoxin VacA produced by Helicobacter pylori [9], as well as a functional receptor for interleukin-34 [10], suggesting that it acts as a functional binding partner for several soluble molecules.
We have recently shown that RPTPβ/ζ-induced, c-Src-mediated β3 Tyr773 phosphorylation is also required for PTN-induced cell surface nucleolin (NCL) localization [11]. NCL is over-expressed on the plasma membrane of cancer and activated endothelial cells and has been shown to play critical roles in the modulation of tumorigenesis and angiogenesis through its interaction with a variety of ligands, among which tumor homing peptide F3, endostatin, P-selectin and PTN [12]. VEGF165 induces NCL localization on the surface of endothelial cells and this effect is considered important for its angiogenic actions [13,14]; however, the receptors and pathways involved have not been elucidated.
In the present work, we explored the possibility that RPTPβ/ζ is involved in the stimulatory effect of VEGF165 on endothelial cell signaling leading to cell migration. Our data show that VEGF165 directly interacts with RPTPβ/ζ to induce c-Src-mediated β3 Tyr773 phosphorylation. The latter is required for both cell surface NCL localization and increased interaction of ανβ3 with VEGFR2, leading to VEGF165-induced endothelial cell migration.
Results and discussion
Phosphorylation of β3 Tyr773 is required for VEGF165-induced cell migration and cell surface NCL localization
It has been shown that phosphorylation of β3 cytoplasmic Tyr 773 and 785 in response to VEGF165 plays a role in endothelial cell migration [2]. In order to determine which of the two Tyr is responsible for VEGF165-induced cell migration, we used CHO cells that express VEGFR2 (Figure 1A), RPTPβ/ζ and αν [8,11], but do not express β3 and are mock-transfected or stably transfected to over-express wild-type β3 or β3 in which Tyr773 and/or Tyr785 are mutated to Phe [11]. VEGF165 induced migration of CHO cells over-expressing wild type β3 or β3Y785F, but had no effect on CHO cells over-expressing β3Y773F or β3Y773F/Y785F (Figure 1B), suggesting that β3 Tyr773 is important for VEGF165-induced cell migration. In the same line and similarly to what we have recently shown for PTN [11], VEGF165-induced cell surface NCL localization was only observed in CHO cells over-expressing wild type-β3 or β3Y785F, while in cells over-expressing β3Y773F, NCL remained restricted in the cell nucleus, suggesting that β3 Tyr773 but not Tyr785 phosphorylation is important for VEGF165-induced cell surface NCL localization (Figure 1C). Since RPTPβ/ζ is involved in PTN-induced β3 Tyr773 phosphorylation and cell surface NCL localization [8,11], these data lead to the hypothesis that RPTPβ/ζ may also be involved in VEGF165-induced signaling that leads to endothelial cell migration.

Phosphorylation of β 3 Tyr773 is required for VEGF 165 -induced cell migration and cell surface NCL localization. (A) Protein extracts of CHO cells were analysed for expression of VEGFR2. HUVEC were used as a positive control and β-actin as a loading control. (B) Effect of VEGF165 (10 ng/ml) on CHO cell migration. Data are from five independent experiments and are expressed as mean ± s.e.m. percentage change in number of migrating cells compared with the corresponding non stimulated cells (set as default 100). (C) Immunofluorescence images stained for NCL (green) and nucleus (blue) in serum starved CHO cells treated with VEGF165 (10 ng/ml) for 5 h at 37°C. Vector, cells transfected with the plasmid vector; wtβ3, cells over-expressing wild-type β3; β3Y773F, cells over-expressing β3Y773F; β3Y785F, cells over-expressing β3Y785F; β3Y773F/Y785F, cells over-expressing double mutant β3Y773F/Y785F.
RPTPβ/ζ plays a role in VEGF165-induced endothelial cell signaling that leads to cell surface NCL localization
Since RPTPβ/ζ is responsible for PTN-induced β3 Tyr773 phosphorylation through dephosphorylation and activation of c-Src in human umbilical vein endothelial cells (HUVEC) [8], we examined whether RPTPβ/ζ also affects VEGF165-induced c-Src activation and β3 Tyr773 phosphorylation. Down-regulation of RPTPβ/ζ expression by two different siRNAs abolished VEGF165-induced dephosphorylation of c-Src at Tyr530 (Figure 2A), suggesting that RPTPβ/ζ may be the missing link for c-Src activation upon stimulation of endothelial cells with VEGF165. Indeed, the increase in dephosphorylated c-Src Tyr530 was mirrored by higher phosphorylation of c-Src at Tyr419 (Figure 2A), verifying activation of c-Src. Interestingly, inhibition of VEGFR2 tyrosine kinase activity by the selective inhibitor SU1498 or inhibition of VEGF-VEGFR2 interaction by bevacizumab, did not affect VEGF165-induced c-Src Tyr530 dephosphorylation or c-Src Tyr419 phosphorylation (Additional file 1), suggesting that c-Src activation may be independent of VEGFR2 in these assays. RPTPβ/ζ directly interacts with c-Src [6], an observation that is in line with a role for RPTPβ/ζ in c-Src activation. Down-regulation of RPTPβ/ζ expression also abolished VEGF165-induced β3 Tyr773 phosphorylation (Figure 2B), suggesting that RPTPβ/ζ may be involved in VEGF165-induced signaling related to cell surface NCL localization, as has been previously described for PTN [11]. Indeed, down-regulation of RPTPβ/ζ expression abolished VEGF165-induced cell surface NCL localization (Figure 2C). Although it has been known for several years that VEGF165 induces cell surface NCL localization [13], which is required for VEGF165-induced cell migration [14], the receptor/pathway involved was unknown up to date. The observation that VEGF165-induced cell surface NCL localization depends on RPTPβ/ζ suggests a role for RPTPβ/ζ in the angiogenic effects of VEGF165.

RPTPβ/ζ is required for VEGF 165 -induced cell surface NCL localization. Serum starved HUVEC were treated with VEGF165 (10 ng/ml) for 10 min. Cell lysates were analyzed by Western blot for non Tyr530 phosphorylated (npc-Src), Tyr419 phosphorylated (pc-Src) and total (tc-Src) c-Src (A), as well as for phospho-β3Y773 (pβ3Y773) and total β3 (tβ3) integrin (B). Numbers in brackets denote the average-fold change of the ratio npc-Src:tc-Src, pc-Src:tc-Src or pβ3Y773:tβ3 respectively, compared with the corresponding non stimulated, untransfected cells (set as default 1). (C) Representative immunofluorescence images stained for NCL (green), RPTPβ/ζ (red) and nucleus (blue) from serum starved HUVEC treated with VEGF165 (10 ng/ml) for 5 h at 37°C. (D) Representative immunofluorescence images stained for NCL (green) and nucleus (blue) from serum starved VEGF165-stimulated HUVEC in the presence or absence of inhibitors for c-Src (PP1 10 μΜ), PI3K (wortmannin 100 nM) and ERK½ (U0126 20 nM). Scale bars in C and D correspond to 10 μm. (E) Lysates from serum starved VEGF165-stimulated HUVEC in the presence or absence of PP1 and wortmannin, were analyzed by Western blot for pβ3Y773 and tβ3 integrin. Numbers in brackets denote the average-fold change of the ratio pβ3Y773:tβ3 compared with untreated cells (set as default 1). (F and G) Phosphorylation of PI3K in HUVEC and CHO cells respectively. Data are expressed as mean ± s.e.m percentage change in PI3K compared with the untreated cells (set as default 100). In all cases, data come from three independent experiments. siNeg, HUVEC transfected with a negative control siRNA; siRPTPβ/ζ1, HUVEC transfected with siRPTPβ/ζ#1; siRPTPβ/ζ2, HUVEC transfected with si RPTPβ/ζ#2; vector, CHO cells transfected with the plasmid vector; wtβ3, CHO cells over-expressing wild-type β3; β3Y773F, CHO cells over-expressing β3Y773F; β3Y785F, CHO cells over-expressing β3Y785F.
To further investigate the involvement of RPTPβ/ζ in VEGF actions, we studied the role of several signaling molecules known to be activated by both VEGF165 and RPTPβ/ζ on cell surface NCL localization. Inhibition of c-Src and phosphatidylinositol 3-kinase (PI3K) abolished VEGF165-induced cell surface NCL localization, while inhibition of ERK1/2 had no effect (Figure 2D). In order to investigate whether PI3K lays up- or downstream of ανβ3, the effect of PI3K inhibition on VEGF165-induced β3 Tyr773 phosphorylation was studied. The c-Src inhibitor PP1 was used as a positive control, since c-Src is known to lay upstream of β3 Tyr773 phosphorylation [2,8,11]. The p38 inhibitor (SB203580 10 μΜ, BioSource Europe, Nivelles, Belgium) was used as a negative control, since it has been previously shown not to affect RPTPβ/ζ-mediated β3 Tyr773 phosphorylation and PI3K activation [11]. PI3K inhibition did not affect VEGF165-induced β3 Tyr773 phosphorylation (Figure 2E), suggesting that it lays downstream of ανβ3. By using an ELISA for activated PI3K in HUVEC, it was found that inhibition of c-Src abolished VEGF165-induced PI3K activation (Figure 2F). Using the same assay in CHO cells over-expressing wild-type β3, β3Y773F or β3Y785F, it was found that VEGF165 significantly induced PI3K activation in CHO cells over-expressing wild-type β3 or β3Y785F, while it had no effect in cells over-expressing β3Y773F (Figure 2G), suggesting that PI3K lays downstream of ανβ3 and requires β3 Tyr773 phosphorylation. This pathway resembles the one we have recently shown for PTN-induced cell surface NCL localization [11], further supporting the notion that RPTPβ/ζ mediates this effect of VEGF165. Similarly to what has been previously discussed for PTN, it remains unclear how PI3K affects cell surface NCL localization. Co-immunoprecipitation of NCL with PI3K [15,16] favors a direct regulation of NCL by PI3K and one possibility is by regulating the trafficking of exocytotic vesicles. NCL has been detected in cytoplasmic vesicles fused with the plasma membrane [17], while inhibition of PI3K interferes with the trafficking of such exocytotic vesicles, affecting the number of several receptors on the plasma membrane, such as the transferrin receptor [18], the glucose transporter GLUT4 [19] or β integrin [20]. Alternatively, PI3K may indirectly regulate cell surface NCL localization, by regulating recruitment of proteins containing pleckstrin homology domains onto the cell membrane [21]. NCL does not possess such sequences but may act as a ligand for proteins containing pleckstrin homology domains through its acidic motifs [22].
RPTPβ/ζ plays a role in VEGF165-induced interaction of VEGFR2 with ανβ3
Based on the literature that c-Src–mediated phosphorylation of β3 essentially regulates VEGF165-induced interaction of ανβ3 with VEGFR2 in endothelial cells [2] and our observation that RPTPβ/ζ is required for VEGF165-induced c-Src activation and β3 Tyr773 phosphorylation (Figure 2), we tested the hypothesis that RPTPβ/ζ may have a role in the interaction of ανβ3 with VEGFR2. Down-regulation of RPTPβ/ζ expression by siRNA abolished the increased interaction of ανβ3 with VEGFR2 induced by VEGF165, as evidenced by both immunoprecipitation/Western blot (Figure 3A) and proximity ligation assays (PLA) (Figure 3B). These data also show involvement of RPTPβ/ζ in VEGF-induced endothelial cell migration.

RPTPβ/ζ regulates VEGF 165 -induced VEGFR2-α ν β 3 interaction. Down-regulation of RPTPβ/ζ expression by siRNA was followed by treatment of serum-starved HUVEC with VEGF165 (10 ng/ml) for 10 min. (A) Cells lysates were immunoprecipitated for β3 and analyzed by Western blot for the presence of VEGFR2 and β3. (B) Formation of β3-VEGFR2 complexes as evidenced by in situ PLA. The box plots indicate the median, mean and range of the detected signals (n = 8 image fields with ~4 cells per image per sample type, each sample run in duplicate). Scale bar corresponds to 10 μm. siNeg, HUVEC transfected with a negative control siRNA; siRPTPβ/ζ, HUVEC transfected with siRPTPβ/ζ#1.
RPTPβ/ζ plays a role in VEGF165-induced endothelial cell migration
Since RPTPβ/ζ affects VEGF165-induced signaling related to cell migration, we tested whether it also affects VEGF165-induced endothelial cell migration. Down-regulation of RPTPβ/ζ expression by siRNA abolished VEGF165-induced HUVEC migration (Figure 4), highlighting a role for RPTPβ/ζ in the angiogenic effects of VEGF165.

RPTPβ/ζ is required for VEGF 165 -induced endothelial cell migration. Effect of VEGF165 on HUVEC migration after down-regulation of RPTPβ/ζ expression by two different siRNAs. Data are from five independent experiments and are expressed as mean ± s.e.m. percentage change in number of migrating cells compared with the non stimulated untransfected cells (set as default 100). Untrasfected, untransfected HUVEC; siNeg, HUVEC transfected with a negative control siRNA; siRPTPβ/ζ1, HUVEC transfected with siRPTPβ/ζ#1; siRPTPβ/ζ2, HUVEC transfected with si RPTPβ/ζ#2. The Western blot on top shows effective down-regulation of RPTPβ/ζ by both siRNA sequences used.
VEGF directly interacts with RPTPβ/ζ
It has been previously shown that RPTPβ/ζ interacts with both ανβ3 and NCL in HUVEC [8,11], both of which are involved in VEGF165-induced endothelial cell migration [2,3,13,14]. Since VEGFR2 is also required for VEGF165-induced endothelial cell migration [1], we tested whether RPTPβ/ζ interacts with VEGFR2. By performing immunoprecipitation/Western blot and PLA assays, we found that VEGFR2 does not associate with RPTPβ/ζ (Figure 5A). We then tested the possibility that VEGF directly associates with RPTPβ/ζ by performing PLA assays in HUVEC, which express endogenous levels of VEGF (Additional file 2). As shown in Figure 5B, endogenous VEGF formed complexes with RPTPβ/ζ, suggesting a direct interaction between the two molecules. Interestingly, addition of exogenous VEGF165 to HUVEC, which led to increased VEGF immunostaining (Additional file 2), increased VEGF-RPTPβ/ζ PLA signals (Figure 5B), verifying the specificity of the signal. In these assays, the interaction of VEGF with VEGFR2 was used as positive control. Interaction of VEGF with RPTPβ/ζ was also observed in human glioblastoma U87MG cells, by using a combination of immunoprecipitation/Western blot, double immunofluorescence and PLA assays. These cells express higher levels of endogenous VEGF compared with HUVEC and the PLA signals showing interaction with RPTPβ/ζ were also higher than those observed in HUVEC (Additional file 2). Interestingly, VEGF was not found to interact with ανβ3 (Additional file 3).

VEGF directly interacts with RPTPβ/ζ in a VEGFR-independent manner. (A) Serum-starved untreated or VEGF165-stimulated HUVEC lysates were immunoprecipitated for VEGFR2 and analyzed by Western blot for the presence of RPTPβ/ζ or VEGFR2 (up). No direct interaction between VEGFR2 with RPTPβ/ζ was observed by performing in situ PLA in HUVEC (down). The RPTPβ/ζ-ανβ3 interaction was used as a positive control. Data are from two independent experiments. (B) Formation of VEGF-RPTPβ/ζ complexes as evidenced by in situ PLA in HUVEC, untreated or after addition of exogenous VEGF165 (10 ng/ml) at 24 h. The VEGF-VEGFR2 interaction was used as a positive control. Data are from five independent experiments. (C) Formation of VEGF-RPTPβ/ζ complexes as evidenced by in situ PLA in HUVEC in the absence or the presence of bevacizumab (250 μg/ml). The box plots in B and C indicate the median, mean and range of the detected signals (n > 20 image fields with ~4 cells per image per sample type, each sample run at least in duplicate) from three independent experiments. (D) Immunofluorescence images stained for NCL (green) and nucleus (blue) in serum starved HUVEC treated for 5 h at 37°C with VEGF165 (10 ng/ml) in the presence or the absence of bevacizumab (250 μg/ml). Representative pictures from three independent experiments. Scale bars in all cases correspond to 10 μm.
Bevacizumab used at a concentration that inhibits VEGF-induced cell migration and binding to both VEGFR1 and VEGFR2 [23], did not affect interaction of VEGF with RPTPβ/ζ (Figure 5C), suggesting that this interaction involves a different region on the VEGF molecule than the VEGFR binding site. Supporting this notion, bevacizumab did not inhibit VEGF165-induced cell surface NCL localization (Figure 5D), which is also mediated by RPTPβ/ζ as discussed above. Bevacizumab at the same concentration significantly inhibited interaction of VEGF with VEGFR2 (Additional file 4). These data clearly indicate that although binding to VEGFR2 is essential for VEGF-induced endothelial cell migration [2], some of the angiogenic actions of VEGF, such as cell surface NCL localization, are not inhibited by bevacizumab. Taking into account that angiogenic factors, such as PTN, hepatocyte growth factor and even VEGF itself act through cell surface NCL [12], the lack of effect of bevacizumab on VEGF165-induced cell surface NCL localization may explain at least some of the cases of resistance development to bevacizumab, e.g. in glioblastomas, where classical VEGF signaling through VEGFRs has been found to remain inhibited [24]. Moreover, our data provide a mechanistic support to the notion that RPTPβ/ζ is a valuable target for glioblastoma therapies [25,26].
There are two transmembrane isoforms of RPTPβ/ζ, the long isoform that has been described as a CS proteoglycan, and the short isoform considered a glycoprotein. Up to date, it is still unclear which of the two RPTPβ/ζ isoforms are responsible for each of its actions and it is not known whether glycosylation of these isoforms differs among different types of cells. Efforts to identify its CS glycanation suggest that it is regulated both developmentally and in different pathophysiological situations [27] and our unpublished observations based on Western blot analyses of RPTPβ/ζ expressed in different cell types support the notion of a cell-type specific RPTPβ/ζ glycosylation. PTN binding to RPTPβ/ζ involves both low (K d = 3 nM) and high (K d = 0.25 nM) affinity binding sites [28], which represent more than one sites of interaction that involve both the protein core and the CS chains of the receptor [5]. Oversulfation of CS is essential for PTN affinity and PTN-mediated functions [29-32], which is in line with our observation that CS-E inhibits interaction of PTN with RPTPβ/ζ in both HUVEC and U87MG cells (Additional file 5). It should be noted, however, that although inhibition in U87MG cells was at the level of 70% (total signals per cell in control: 23 ± 7 and in CS-E treated cells: 8 ± 1), in HUVEC it was smaller, at the level of 50% (total signals per cell in control: 6 ± 0.4 and in CS-E treated cells: 3 ± 0.3).
Since VEGF has been shown to be able to bind to CS-E similarly to heparan sulphate, but does not bind CS-C or CS-A [33], we investigated whether CS-Ε also inhibits VEGF interaction with RPTPβ/ζ. CS-E decreased interaction of VEGF with RPTPβ/ζ in U87MG cells (Additional file 6), but had no effect on HUVEC (Figure 6A), in line with the observation that it did not affect VEGF165-induced cell surface NCL localization (Figure 6B). This difference could be explained by the hypothesis that cell surface NCL localization depends on the interaction of VEGF with the short, non-proteoglycan isoform of RPTPβ/ζ. Interestingly and in favor of such a possibility, in U87MG cells that express higher amounts of endogenous VEGF than HUVEC, we performed IP/Western blot assays and observed that CS-E inhibits interaction of both PTN and VEGF with the long, but not the short RPTPβ/ζ isoform (Additional file 6). These data suggest that interaction of VEGF (and PTN) with the short RPTPβ/ζ isoform may not involve CS-E chains. On the other hand, CS-E abolished VEGF165-induced HUVEC migration (Figure 6C), an effect that may involve a different cell surface molecule or parallel signaling pathways that are important for cell migration. This point is under further investigation.

Effect of CS-E on VEGF 165 -induced endothelial cell signaling and migration. (A) Formation of VEGF-RPTPβ/ζ complexes as evidenced by in situ PLA in HUVEC in the absence or the presence of CS-E II (100 ng/ml). The box plots indicate the median, mean and range of the detected signals (n > 20 image fields with ~4 cells per image per sample type, each sample run at least in duplicate) from three independent experiments. (B) Immunofluorescence images stained for NCL (green) and nucleus (blue) in serum starved HUVEC treated for 5 h at 37°C with VEGF165 (10 ng/ml) in the absence or the presence of CS-E II (100 ng/ml). Representative pictures from two independent experiments. (C) Effect of CS-E I and II (both at 100 ng/ml) on VEGF165-induced HUVEC migration. Data are from three independent experiments and are expressed as mean ± s.e.m. percentage change in number of migrating cells compared with the non stimulated untransfected cells (set as default 100). (D) Immunofluorescence images stained for NCL (green) and nucleus (blue) in serum starved HUVEC treated for 5 h at 37°C with VEGF121 (10 ng/ml). Representative pictures from two independent experiments. Scale bars in all cases correspond to 10 μm.
The idea that the interaction of VEGF with RPTPβ/ζ does not involve its heparin-binding properties is further supported by the observation that VEGF121, which induces HUVEC migration but does not contain the heparin-binding site of VEGF [34], also induces cell surface NCL localization (Figure 6D). Collectively, our data suggest that interaction of VEGF with RPTPβ/ζ may involve a part of the molecule that is distinct from those involved in VEGFR or glycosaminoglycan binding. This notion is similar to the recent finding that the high affinity neuropilin-1 binding of VEGF-A involves the exon 8-encoded C-terminal Arg [35]. Tuftsin, a naturally occurring TKPR peptide with sequence similarity to the sequence coded by exon 8 of VEGF, blocks VEGF165-induced autophosphorylation of VEGFR2, without inhibiting VEGF binding to VEGFR2 [36]. Taking into account that PTN binding to RPTPβ/ζ involves PTN's carboxy terminal region that is rich in basic amino acids [37], and that PTN inhibits VEGF binding to RPTPβ/ζ (see below), one can speculate that binding of VEGF to RPTPβ/ζ might be mediated by its exon 8-encoded sequence.
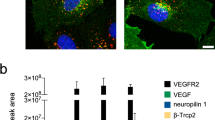
Finally, we found that the interaction of VEGF with RPTPβ/ζ was decreased in the presence of PTN in both HUVEC (Figure 7A) and U87MG cells (Additional file 7), suggesting that these two growth factors share a common binding site on RPTPβ/ζ. This is further supported by the observation that the PTN-RPTPβ/ζ interaction was also decreased in the presence of exogenous VEGF165 (Figure 7B). It has been previously suggested that besides the CS chains, the core RPTPβ/ζ protein is also involved in binding to PTN [28,38], which may also be the site of interaction of the short RPTPβ/ζ transmembrane isoform with VEGF, as discussed above.

PTN and VEGF compete for binding to RPTPβ/ζ. (A) Formation of VEGF-RPTPβ/ζ complexes as evidenced by in situ PLA in HUVEC in the absence or the presence of exogenous PTN (100 ng/ml). (Β) Formation of PTN-RPTPβ/ζ complexes as evidenced by in situ PLA in HUVEC in the absence or the presence of exogenous VEGF165 (10 ng/ml). The box plots in A and B indicate the median, mean and range of the detected signals (n > 20 image fields with ~4 cells per image per sample type, each sample run at least in duplicate) from three independent experiments in each case. (C) Effect of PTN (100 ng/ml) on VEGF165-induced HUVEC migration. Data are from five independent experiments and are expressed as mean ± s.e.m. percentage change in number of migrating cells compared with the non stimulated untransfected cells (control, set as default 100). Asterisks denote a statistically significant difference from control. ***P < 0.001.
PTN stimulates human endothelial cell migration by approximately 40% [6,8,11], an effect that is smaller than that of other growth factors, such as VEGF. Interestingly, PTN decreased VEGF165-induced HUVEC migration to the levels of its own, smaller stimulatory effect (Figure 7C), in agreement with a previous study showing that PTN did not abolish but decreased VEGF165-induced endothelial cell infiltration of matrigel in vivo to the levels of its own stimulation [39]. These data favor the notion that PTN and VEGF compete for binding to RPTPβ/ζ, as discussed above, and highlight a possible role for PTN as a regulator of angiogenesis, by limiting the aberrant effect of VEGF, while it still induces a smaller, significant stimulatory effect.
It is important to note that although RPTPβ/ζ is required for cell migration induced by PTN [6] and VEGF165 (present study), it is not sufficient by itself to induce cell migration, based on our previous data showing that midkine [8] or PTN112–136 [40], which lead to c-Src Tyr530 dephosphorylation and β3 Τyr773 phosphorylation through RPTPβ/ζ, do not induce endothelial cell migration. It seems that other receptors/signaling pathways are activated in parallel with the RPTPβ/ζ/c-Src/ανβ3 pathway to complementarily induce cell migration. Such a pathway for VEGF165 involves at least VEGFR2 and the cross-talk of signaling molecules activated by VEGF165 through RPTPβ/ζ, ανβ3 and VEGFR2 or other cell surface binding molecules in different cell types is under further investigation.
Conclusion
RPTPβ/ζ is a receptor-type protein tyrosine phosphatase expressed in several types of cancer and involved in cell migration, cancer progression and metastasis [5,27]. The current study identifies RPTPβ/ζ as a novel cell membrane binding molecule for VEGF165, which regulates c-Src-mediated β3 Tyr773 phosphorylation and interaction with VEGFR2, cell surface NCL localization through PI3K activation and endothelial cell migration (Figure 8). These data are of high significance, especially taking into account that the interaction of VEGF with RPTPβ/ζ and the downstream angiogenic VEGF actions, such as cell surface NCL localization, do not seem to be affected by existing anti-VEGF drugs, such as bevacizumab, and may explain the resistance developed by tumor types, e.g. glioblastoma, in such therapies [24,41]. They thus warrant exploitation of RPTPβ/ζ for the possible development of alternative or additive anti-angiogenic therapies, especially in cases where resistance develops.

Schematic representation of the proposed mechanism that involves RPTPβ/ζ and leads to increased α ν β 3 -VEGFR2 interaction, cell surface NCL localization and stimulation of cell migration by VEGF. Binding of VEGF165 to RPTPβ/ζ on the surface of endothelial or cancer cells leads to c-Src activation, β3 Tyr773 phosphorylation and increased interaction of ανβ3 with VEGFR2, as well as PI3K activation and translocation of NCL from the nucleus to the cell membrane. Both are required for VEGF165-induced endothelial cell migration. For more details see text.
Methods
Materials
Human recombinant VEGF165 was prepared as previously described [39]. VEGF121 was purchased from RELIATech GmbH (Wolfenbüttel, Germany). Bevacizumab (AVASTIN) was from Roche Applied Science (Indianapolis, IN, USA). Human recombinant PTN was from PeproTech, Inc. (Rocky Hill, NJ, USA) or prepared as previously described [39]. PTN from both sources was equally active in all cases. CS-E was either from Seikagaku Corporation (Tokyo, Japan) (CS-E I), or prepared from a crude preparation of squid cartilage CS (purchased from Yantai Changsen Chemical Co., Ltd., Shandong, China) (CS-E II), both sources being equally active. PP1, wortmannin and U0126 were from TOCRIS (Minneapolis, MN, USA) and SU1498 was from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). All secondary horseradish peroxidase-conjugated antibodies were from Cell Signaling Technology Inc. (Beverly, MA, USA). Human IgG and other reagents were from Sigma (St. Louis, MO, USA).
Cell culture
HUVEC, human glioma U87MG cells and CHO cells expressing αν but deficient in endogenous β3 integrin or stably transfected to express wild type β3, β3Y773F, β3Y785F or β3Y773F/Y785F were cultured as previously described [8,11]. Cell culture reagents were from BiochromKG (Seromed, Germany). All cultures were maintained at 37°C, 5% CO2, and 100% humidity. When cells reached 70-80% confluence, they were serum starved for 16 h (where indicated) before performing migration assays, lysed for immunoprecipitation/Western blot assays or fixed for immunofluorescence assays and in situ PLA.
Immunofluorescence
Cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS) pH 7.4 for 10 min and permeabilized with 0.1% Triton in PBS for 15 min. After being washed 3 times with PBS, the cells were blocked with PBS containing 3% BSA and 10% fetal bovine serum (FBS) for 1 h at room temperature. Cells were stained with the following primary antibodies: rabbit anti-VEGF (1:250; Santa Cruz Biotechnology Inc.), mouse anti-RPTPβ/ζ (1:250; BD Biosciences, San Diego, CA, USA), mouse anti-ανβ3 (1:500; Merck Millipore, Darmstadt, Germany), and rabbit anti-NCL (1:1,000, Sigma). Cells were then incubated with fluorescent Alexa secondary antibodies (1:500; Molecular Probes, Carlsbad, CA, USA). Nuclei were stained with Draq5 (Biostatus Limited, Leicestershire, UK). Cells were mounted with Mowiol 4–88 (Merck Millipore) and visualized at room temperature with Leica SP5 (X63 objective with a numerical aperture of 1.4) confocal microscope.
In situ PLA
For detection of protein-protein interactions, in situ PLA was performed. The components used (Sigma) were as follows: anti-mouse PLA plus probe, anti-rabbit PLA minus probe and Detection Reagents Orange. HUVEC or U87MG cells were grown on μ-Chamber 12 well on glass slides (Ibidi©, Martinsried, Germany). After reaching 80% confluence or after appropriate treatment of cells, the assay was performed according to the manufacturer’s instructions. Briefly, after fixation and blocking, cells were incubated with the primary antibodies: mouse anti-VEGF (1:250), rabbit anti-VEGF (1:250), rabbit anti-Flk-1 (1:250), mouse anti-Flk-1 (1:250), mouse anti-NCL (1:50), goat anti-RPTPβ/ζ (1:250) (all from Santa Cruz Biotechnology Inc.), mouse anti-ανβ3 (1:500; Merck Millipore), mouse anti-PTN (1:500; Abnova, Heidelberg, Germany) and mouse anti-RPTPβ/ζ (1:250; BD Biosciences). Subsequently, cells were incubated with secondary antibodies conjugated with oligonucleotides. After hybridization and ligation of the oligonucleotides, the DNA was amplified. A detection mixture detected the amplicons, resulting in red fluorescence signals. Nuclei were counterstained with Draq5; cells were mounted with Mowiol 4–88 and visualized at room temperature with Leica SP5 confocal microscope.
Quantification of in situ PLA signals
Estimation of nuclei and cytoplasm size was performed using the Duolink ImageTool software (Olink Bioscience). In order to calculate the total number of spots per cell, an algorithmic procedure was developed and implemented in the Matlab environment (The MathWorks Inc., Massachusetts, USA). “I” denotes an immunofluorescence image of m × n size. Such an image can be seen as a “stack” of three matrices of the same size; representing the red, green and blue values for each pixel indicated as IR, IG, IB for the three color bands, respectively. For every pixel at position (i, j) correspond three values; IR(i, j), IG(i, j), IB(i, j), ranging from 0 to 255. Image pre-processing was performed to identify pixels of red intensities less than a determined threshold value θ, in order to exclude background noise. Several values were tested and evaluated and finally θ was set to 50. In order to detect a red dot, the algorithm searches the red band (i.e., the IR matrix) for a group of at least c connected pixels. The value of the parameter c, which controls the minimum number of pixels that constitute a dot, c was set to 10. Moreover, in the case of overlapping dots, large groups of pixels were identified. In such cases, the true number of dots was calculated as [n /100] since a typical dot contains approximately 100 pixels, where n is the total number of pixels of the initial group and the operator [α] is used to round the number α into its nearest integer.
RNA interference
Cells were grown to 50% confluence in medium without antibiotics. Transfection was performed in serum-free medium for 4 h using annealed RNA for RPTPβ/ζ (siRPTPβ/ζ#1, VBC Biotech Services, Vienna, Austria), as previously described [6]. Another siRNA sequence for RPTPβ/ζ (siRPTPβ/ζ#2, Hs_PTPRZ1_1 FlexiTube siRNA, Qiagen GmbH, Germany) was also used as a control for off-target effects. JetSI-ENDO (Polyplus Transfection, Illkirch, France) or Lipofectamine® RNAiMAX (Life Technologies) were used as transfection reagents. Double-stranded negative control siRNA (Ambion, Austin, TX, USA) was used in all experiments. Cells were incubated for another 48 h in serum-containing medium and lysed, serum starved or fixed before further experiments.
Migration assays
Migration assays were performed as previously described [8,11] in 24-well microchemotaxis chambers (Corning, Inc., Lowell, MA, USA) using uncoated polycarbonate membranes with 8 μm pores. Serum-starved cells were harvested, resuspended at a concentration of 105 cells/0.1 ml in serum-free medium containing 0.25% bovine serum albumin (BSA) and loaded in the upper chamber. The bottom chamber was filled with 0.6 ml of serum-free medium containing 0.25% BSA and the tested substances. Cells were incubated for 4 h at 37°C. After completion of the incubation, filters were fixed and stained with 0.33% toluidine blue solution. The cells that migrated through the filters were quantified by counting the entire area of each filter, using a grid and a microscope with a X20 objective (Optech Microscope Services Ltd., Thames, UK).
PI3K p85 ELISA
The levels of total and phosphorylated PI3K p85 were quantified using Fast Activated Cell-based ELISA assays (Active Motif, Carlsbad, CA, USA) according to the manufacturer's instructions. Briefly, cells were cultured in 96-well plates one day prior to manipulation. Serum starved CHO cells or HUVEC were treated with 10 ng/ml VEGF165 for 10 min (in the presence or absence of inhibitors of signaling molecules where appropriate), fixed and incubated with anti–phospho and anti-total p85 antibodies.
Immunoprecipitation assay
Cells were lysed with RIPA buffer, as previously described [8]. Three mg of total protein were incubated with primary antibody for 16 h at 4°C under continuous agitation. The primary antibodies used were: mouse anti-VEGF (3 μg), mouse anti-Flk-1 (3 μg), goat anti-RPTPβ/ζ (3 μg), goat anti-PTN (3 μg) (Santa Cruz Biotechnology Inc.) and goat anti-β3 (1.5 μg; Merck Millipore). Protein A- and protein G-agarose beads (Merck Millipore) were added, samples were further incubated for 2 h at 4°C, and beads with bound proteins were collected by centrifugation (5,000 g for 5 min at 4°C) and washed twice with ice-cold PBS pH 7.4. Immunoprecipitated proteins were resuspended in SDS loading buffer and analyzed by Western blot.
Western blot analysis
Proteins were analyzed by SDS-PAGE and transferred to Immobilon P membranes. Blocking was performed by incubating the membranes with Tris-buffered saline (TBS) pH 7.4 with 0.05% Tween (TBS-T), containing either 5% nonfat dry milk or 3% BSA. Membranes were incubated with primary antibodies for 16 h at 4°C under continuous agitation, washed 3 times with TBS-T, and incubated with secondary antibodies for 1 h at room temperature. Primary antibodies used were mouse anti-Flk-1 (1:500), goat anti-β3 (1:500), mouse anti-VEGF (1:500), rabbit anti-phospho-β3(Y773) (1:1,000; Santa Cruz Biotechnology Inc.), mouse anti-RPTPβ/ζ (1:500; BD Biosciences), rabbit anti-c-Src (1:1,000; Merck Millipore), rabbit anti-phospho-c-Src(Y418) (1:1,000; Acris Antibodies GmbH) and rabbit anti-non-phospho-c-Src (1:1,000; Cell Signaling Technology Inc.). Detection of immunoreactive bands was performed using the enhanced chemiluminescence detection kit (Pierce Biotechnology, Rockford, IL, USA). Protein levels were quantified using the ImagePC image analysis software (Scion Corp., Frederick, MD, USA).
Statistical analysis
Results are expressed as mean ± s.e.m. or by using box plots, where the box is determined by the 25th and 75th percentiles, the whiskers are determined by the 5th and 95th percentiles, the line in the box marks the median and the empty square in the box marks the mean. Outliers have been plotted as individual points. Where applicable, variability between the results from each group and the corresponding control was determined by unpaired t test.
Abbreviations
- BSA:
-
Bovine serum albumin
- CS:
-
Chondroitin sulphate
- ELISA:
-
Enzyme-linked immunoassay
- FBS:
-
Fetal bovine serum
- NCL:
-
Nucleolin
- PBS:
-
Phosphate buffered saline
- PI3K:
-
Phosphatidylinositol 3-kinase
- PLA:
-
Proximity ligation assay
- PTN:
-
Pleiotrophin
- RPTPβ/ζ:
-
Receptor protein tyrosine phosphatase beta/zeta
- TBS:
-
Tris-buffered saline
- VEGF:
-
Vascular endothelial growth factor
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
References
Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012;2:a006502.
Mahabeleshwar GH, Feng W, Reddy K, Plow EF, Byzova TV. Mechanisms of integrin-vascular endothelial growth factor receptor cross-activation in angiogenesis. Circ Res. 2007;101:570–80.
Somanath PR, Malinin NL, Byzova TV. Cooperation between integrin alphavbeta3 and VEGFR2 in angiogenesis. Angiogenesis. 2009;12:177–85.
Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008;1784:56–65.
Koutsioumpa M, Papadimitriou E: PG receptors with phosphatase action in cancer and angiogenesis. In Extracellular Matrix: Pathobiology and Signalling. Edited by Karamanos N. Berlin/Boston: Walter de Gruyter GmbH and Co KG; 2012:813–823.
Polykratis A, Katsoris P, Courty J, Papadimitriou E. Characterization of heparin affin regulatory peptide signaling in human endothelial cells. J Biol Chem. 2005;280:22454–61.
Papadimitriou E, Mikelis C, Lampropoulou E, Koutsioumpa M, Theochari K, Tsirmoula S, et al. Roles of pleiotrophin in tumor growth and angiogenesis. Eur Cytokine Netw. 2009;20:180–90.
Mikelis C, Sfaelou E, Koutsioumpa M, Kieffer N, Papadimitriou E. Integrin alpha(v)beta(3) is a pleiotrophin receptor required for pleiotrophin-induced endothelial cell migration through receptor protein tyrosine phosphatase beta/zeta. FASEB J. 2009;23:1459–69.
Fujikawa A, Shirasaka D, Yamamoto S, Ota H, Yahiro K, Fukada M, et al. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat Genet. 2003;33:375–81.
Nandi S, Cioce M, Yeung YG, Nieves E, Tesfa L, Lin H, et al. Receptor-type protein-tyrosine phosphatase ζ is a functional receptor for interleukin-34. J Biol Chem. 2013;288:21972–86.
Koutsioumpa M, Polytarchou C, Courty J, Zhang Y, Kieffer N, Mikelis C, et al. Interplay between αvβ3 integrin and nucleolin regulates human endothelial and glioma cell migration. J Biol Chem. 2013;288:343–54.
Koutsioumpa M, Papadimitriou E. Cell surface nucleolin as a target for anti-cancer therapies. Recent Pat Anticancer Drug Discov. 2014;9:137–52.
Huang Y, Shi H, Zhou H, Song X, Yuan S, Luo Y. The angiogenic function of nucleolin is mediated by vascular endothelial growth factor and nonmuscle myosin. Blood. 2006;107:3564–71.
Destouches D, El Khoury D, Hamma-Kourbali Y, Krust B, Albanese P, Katsoris P, et al. Suppression of tumor growth and angiogenesis by a specific antagonist of the cell-surface expressed nucleolin. PLoS One. 2008;3:e2518.
Barel M, Le Romancer M, Frade R. Activation of the EBV/C3d receptor (CR2, CD21) on human B lymphocyte surface triggers tyrosine phosphorylation of the 95-kDa nucleolin and its interaction with phosphatidylinositol 3 kinase. J Immunol. 2001;166:3167–73.
Wu DM, Zhang P, Liu RY, Sang YX, Zhou C, Xu GC, et al. Phosphorylation and changes in the distribution of nucleolin promote tumor metastasis via the PI3K/Akt pathway in colorectal carcinoma. FEBS Lett. 2014;588:1921–9.
Hovanessian AG, Puvion-Dutilleul F, Nisole S, Svab J, Perret E, Deng JS, et al. The cell-surface-expressed nucleolin is associated with the actin cytoskeleton. Exp Cell Res. 2000;261:312–28.
Spiro DJ, Boll W, Kirchhausen T, Wessling-Resnick M. Wortmannin alters the transferrin receptor endocytic pathway in vivo and in vitro. Mol Biol Cell. 1996;7:355–67.
Thong FS, Dugani CB, Klip A. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology (Bethesda). 2005;20:271–84.
Abe M, Setoguchi Y, Tanaka T, Awano W, Takahashi K, Ueda R, et al. Membrane protein location-dependent regulation by PI3K (III) and rabenosyn-5 in Drosophila wing cells. PLoS One. 2009;4:e7306.
Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350(Pt 1):1–18.
Burks DJ, Wang J, Towery H, Ishibashi O, Lowe D, Riedel H, et al. IRS pleckstrin homology domains bind to acidic motifs in proteins. J Biol Chem. 1998;273:31061–7.
Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335–45.
Lu KV, Bergers G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013;2:49–65.
Muller S, Kunkel P, Lamszus K, Ulbricht U, Lorente GA, Nelson AM, et al. A role for receptor tyrosine phosphatase ζ in glioma cell migration. Oncogene. 2003;22:6661–8.
Ulbricht U, Eckerich C, Fillbrandt R, Westphal M, Lamszus K. RNA interference targeting protein tyrosine phosphatase ζ/receptor-type protein tyrosine phosphatase β suppresses glioblastoma growth in vitro and in vivo. J Neurochem. 2006;98:1497–506.
Pantazaka E, Papadimitriou E. Chondroitin sulfate-cell membrane effectors as regulators of growth factor-mediated vascular and cancer cell migration. Biochim Biophys Acta. 1840;2014:2643–50.
Maeda N, Nishiwaki T, Shintani T, Hamanaka H, Noda M. 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase ζ/RPTPβ, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM). J Biol Chem. 1996;271:21446–52.
Maeda N, Fukazawa N, Hata T. The binding of chondroitin sulfate to pleiotrophin/heparin-binding growth-associated molecule is regulated by chain length and oversulfated structures. J Biol Chem. 2006;281:4894–902.
Tanaka M, Maeda N, Noda M, Marunouchi T. A chondroitin sulfate proteoglycan PTPζ /RPTPβ regulates the morphogenesis of Purkinje cell dendrites in the developing cerebellum. J Neurosci. 2003;23:2804–14.
Bao X, Muramatsu T, Sugahara K. Demonstration of the pleiotrophin-binding oligosaccharide sequences isolated from chondroitin sulfate/dermatan sulfate hybrid chains of embryonic pig brains. J Biol Chem. 2005;280:35318–28.
Deepa SS, Umehara Y, Higashiyama S, Itoh N, Sugahara K. Specific molecular interactions of oversulfated chondroitin sulfate E with various heparin-binding growth factors. Implications as a physiological binding partner in the brain and other tissues. J Biol Chem. 2002;277:43707–16.
ten Dam GB, van de Westerlo EM, Purushothaman A, Stan RV, Bulten J, Sweep FC, et al. Antibody GD3G7 selected against embryonic glycosaminoglycans defines chondroitin sulfate-E domains highly up-regulated in ovarian cancer and involved in vascular endothelial growth factor binding. Am J Pathol. 2007;171:1324–33.
Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–26.
Parker MW, Xu P, Guo HF, Vander Kooi CW. Mechanism of selective VEGF-A binding by neuropilin-1 reveals a basis for specific ligand inhibition. PLoS One. 2012;7:e49177.
von Wronski MA, Raju N, Pillai R, Bogdan NJ, Marinelli ER, Nanjappan P, et al. Tuftsin binds neuropilin-1 through a sequence similar to that encoded by exon 8 of vascular endothelial growth factor. J Biol Chem. 2006;281:5702–10.
Lu KV, Jong KA, Kim GY, Singh J, Dia EQ, Yoshimoto K, et al. Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem. 2005;280:26953–64.
Bermek O, Diamantopoulou Z, Polykratis A, Dos Santos C, Hamma-Kourbali Y, Burlina F, et al. A basic peptide derived from the HARP C-terminus inhibits anchorage-independent growth of DU145 prostate cancer cells. Exp Cell Res. 2007;313:4041–50.
Heroult M, Bernard-Pierrot I, Delbe J, Hamma-Kourbali Y, Katsoris P, Barritault D, et al. Heparin affin regulatory peptide binds to vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. Oncogene. 2004;23:1745–53.
Mikelis C, Lamprou M, Koutsioumpa M, Koutsioubas AG, Spyranti Z, Zompra AA, et al. A peptide corresponding to the C-terminal region of pleiotrophin inhibits angiogenesis in vivo and in vitro. J Cell Biochem. 2011;112:1532–43.
Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320:130–7.
Acknowledgements
This work was supported by the European Union (European Social Fund – ESF, Heracleitus II (to M. Koutsioumpa - E. Papadimitriou), Thales (to V. Megalooikonomou) and ΙΚΥ Fellowship of Excellence for Postgraduate studies in Greece - Siemens Program (to E. Pantazaka). This work was also supported in part by a Grant-in-Aid for Scientific Research for Challenging Exploratory Research 25670018 (to K. Sugahara) from the Japan Society for the Promotion of Science (JSPS). The authors thank the Advanced Light Microscopy facility of the Medical School, University of Patras for using the Leica SP5 confocal microscope and the PhD candidate Margarita Lamprou for technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MK, EPo and EPap participated in research design. MK and EPa performed PLA assays and EPa, AS and VM contributed analytical tools for PLA signal quantification. MK, EPo, EPa and CT performed immunoprecipitation/Western blot assays. SM and KS prepared CS-E and provided expertise on CS. NK provided stably transfected cell lines and expertise on integrins. JC contributed new analytic tools. MK, EPo, EPa, CT, AS and EPap performed data analysis. All authors contributed to the writing of the manuscript. All authors read and approved the manuscript.
Additional files
Additional file 1:
c-Src activation may be independent of VEGF-VEGFR2 interaction. Serum starved HUVEC were treated with VEGF165 (10 ng/ml) for 10 min in the presence or absence of either the selective VEGFR2 tyrosine kinase inhibitor SU1498 (10 μΜ) or bevacizumab (250 μg/ml). Cell lysates were analyzed by Western blot for non Tyr530 phosphorylated (npc-Src), Tyr419 phosphorylated (pc-Src) and total (tc-Src) c-Src. Numbers in brackets denote the average-fold change of the ratio npc-Src:tc-Src or pc-Src:tc-Src respectively, compared with the corresponding non stimulated, untransfected cells (set as default 1) from three independent experiments.
Additional file 2:
VEGF-RPTPβ/ζ interaction in human glioma U87MG cells. (A) Immunofluorescence images of HUVEC and U87MG cells stained for VEGF (green) and nucleus (blue). Representative pictures from three and two independent experiments respectively. (B) U87MG cell lysates were immunoprecipitated for RPTPβ/ζ or VEGF. Immunoprecipitates were analyzed by Western blot for the presence of VEGF or RPTPβ/ζ, respectively. IgG was used as a negative control. Representative blots from two independent experiments. (C) Immunofluorescence images of U87MG cells stained for VEGF (green), RPTPβ/ζ (red) and nucleus (blue). Representative pictures from two independent experiments. (D) In situ PLA signals were detected as red dots, indicating the direct formation of VEGF- RPTPβ/ζ complexes. Representative pictures from three independent experiments. Scale bars in A, C and D correspond to 10 μm. (E) Formation of VEGF-RPTPβ/ζ complexes as evidenced by in situ PLA in HUVEC and U87MG cells. The box plots indicate the median, mean and range of the detected signals (n > 20 image fields with 4-6 cells per image per sample type, each sample run in duplicate) from four independent experiments.
Additional file 3:
VEGF does not interact with α ν β 3 . (A) Immunofluorescence images of U87MG cells cultured in serum-containing medium and stained for VEGF (green), ανβ3 (red) and nucleus (blue). Representative pictures from two independent experiments. (B) The absence of in situ PLA signals indicates lack of the VEGF-ανβ3 direct interaction in both HUVEC and U87MG cells. The PTN-ανβ3 interaction in both types of cells was used as a positive control. Representative pictures from two independent experiments.
Additional file 4:
Bevacizumab inhibits VEGF-VEGFR2 interaction. Formation of VEGF-VEGFR2 complexes as evidenced by in situ PLA in HUVEC in the absence or the presence of bevacizumab (250 μg/ml). The box plots indicate the median, mean and range of the detected signals (n > 20 image fields with ~4 cells per image per sample type, each sample run at least in duplicate) from two independent experiments. Scale bars in all cases correspond to 10 μm.
Additional file 5:
CS-E inhibits PTN-RPTPβ/ζ interaction in both HUVEC and U87MG cells. Formation of PTN-RPTPβ/ζ complexes as evidenced by in situ PLA in HUVEC (A) and U87MG cells (B) in the absence or presence of CS-E II (100 ng/ml). The box plots indicate the median, mean and range of the detected signals (n = 20 image fields with ~6 cells per image per sample type, each sample run in duplicate) from four (A) and three (B) independent experiments. Scale bars in all cases correspond to 10 μm.
Additional file 6:
CS-E inhibits VEGF-RPTPβ/ζ interaction in U87MG cells. (A) Formation of VEGF-RPTPβ/ζ complexes as evidenced by in situ PLA in U87MG cells in the absence or presence of CS-E II (100 ng/ml). The box plots indicate the median, mean and range of the detected signals (n = 8 image fields with ~4 cells per image per sample type, each sample run in duplicate) from three independent experiments. Scale bar corresponds to 10 μm. (B) U87MG cell lysates were immunoprecipitated for VEGF. Immunoprecipitates were analyzed by Western blot for the presence of RPTPβ/ζ. (C) U87MG cell lysates were immunoprecipitated for PTN. Immunoprecipitates were analyzed by Western blot for the presence of RPTPβ/ζ. In B and C, representative blots from two independent experiments are shown.
Additional file 7:
PTN inhibits VEGF-RPTPβ/ζ interaction in U87MG cells. (A) Formation of VEGF-RPTPβ/ζ complexes as evidenced by in situ PLA in U87MG cells in the absence or presence of PTN (100 ng/ml). The box plots indicate the median, mean and range of the detected signals (n = 6 image fields with ~ 5 cells per image per sample type, each sample run in duplicate). Scale bar corresponds to 10 μm. Data come from three independent experiments. (B) Cell lysates were immunoprecipitated for RPTPβ/ζ. Immunoprecipitates were analyzed by Western blot for the presence of VEGF. IgG was used as a negative control. Representative blots from two independent experiments.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Koutsioumpa, M., Poimenidi, E., Pantazaka, E. et al. Receptor protein tyrosine phosphatase beta/zeta is a functional binding partner for vascular endothelial growth factor. Mol Cancer 14, 19 (2015). https://doi.org/10.1186/s12943-015-0287-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-015-0287-3