
Abstract
Background
Hepatocellular carcinoma (HCC) one of the most common digestive system tumors, threatens the tens of thousands of people with high morbidity and mortality world widely. The purpose of our study was to investigate the related genes of HCC and discover their potential abilities to predict the prognosis of the patients.
Methods
We obtained RNA sequencing data of HCC from The Cancer Genome Atlas (TCGA) database and performed analysis on protein coding genes. Differentially expressed genes (DEGs) were selected. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were conducted to discover biological functions of DEGs. Protein and protein interaction (PPI) was performed to investigate hub genes. In addition, a method of supervised machine learning, recursive feature elimination (RFE) based on random forest (RF) classifier, was used to screen for significant biomarkers. And the basic experiment was conducted by lab, we constructe a clinical patients’ database, and obtained the data and results of immunohistochemistry.
Results
We identified five biomarkers with significantly high expression to predict survival risk of the HCC patients. These prognostic biomarkers included SPC25, NUF2, MCM2, BLM and AURKA. We also defined a risk score model with these biomarkers to identify the patients who is in high risk. In our single-center experiment, 95 pairs of clinical samples were used to explore the expression levels of NUF2 and BLM in HCC. Immunohistochemical staining results showed that NUF2 and BLM were significantly up-regulated in immunohistochemical staining. High expression levels of NUF2 and BLM indicated poor prognosis.
Conclusion
Our investigation provided novel prognostic biomarkers and model in HCC and aimed to improve the understanding of HCC. In the results obtained, we also conducted a part of experiments to verify the theory described earlier, The experimental results did verify our theory.
Similar content being viewed by others
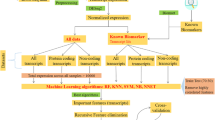


Background
Hepatocellular carcinoma (HCC) is considered to be the most common liver cancer in the world, ranking fifth in men and seventh in women [1]. The development of liver cancer is highly correlated with the infection of hepatitis B virus (HBV) and/or hepatitis C virus (HCV) [2]. Surgical resection is the main treatment for most cases of liver cancer (HCC) and only 30 to 40% of patients with liver cancer can be treated after diagnosis by surgical resection [3]. Therefore, it is important to find an effective and reliable diagnosis of liver cancer that can significantly improve the diagnosis of liver cancer patients.
Carcinogenesis is a multi-step process, which is a change caused by signal pathways triggered by multiple genes, which transforms normal cells into malignant cells [4, 5]. The molecular mechanism of the occurrence and development of HCC are unclear. However, it is considered that, at the beginning of the preneoplastic stage, genetic changes in a few genes and chromosomal loci will slowly accelerate and enhance the transition of hepatocytes from atypical hyperplasia to liver cancer [6]. With the development of Next Generation Sequencing (NGS) technologies, we have located key carcinogenic genes and related oncogenic signaling pathways that play a pivotal role in the initiation and progression of HCC. However, despite the availability of a large amount of public genetic information, effective diagnostic methods are needed to predict the prognosis of HCC.
With a series of changes in biological processes, such as immune regulation, cell cycle, angiogenesis, healing, and auto-swallowing, genetic mutations contribute significantly to tumor formation [7,8,9,10,11]. Differentially expressed genes (DEGs) are involved in changes in signal routing and biological processes during tumor formation. Tracks were not independent in their function, which are linked between tracks. Interfering genes revealed by related pathways are potential biomarkers and therapeutic targets for cancer. Important information about liver cancer can be found in these intersecting genes.
In this study, the HCC gene expression profile data was downloaded from the public database to determine a linear risk score as a survival prediction model based on the HCC interference genes and for identify biomarkers that predict the risk of survival for patients with liver cancer.
Materials and methods
RNA-seq transcriptome data of samples
We downloaded the RNA-sequencing (RNA-seq) expression profiles of HCC from TCGA database (https://cancergenome.nih.gov/, up to Nov.03, 2016), involving 423 samples. These samples contained 373 HCC tumor tissues and 50 normal liver tissues, which were publicly available and open-access. The clinical data of HCC patients were also obtained from TCGA and 369 patients with complete survival data were enrolled in further survival analysis. Data acquired from TCGA database were carried out by the Illumina HiSeq Systems. Data format of sequencing is Counts files.
Gene reannotation
RNA-sequencing data got from the TCGA contained multiple types of RNA, including long non-coding RNA (lncRNA), protein coding genes and pseudogenes. The transcriptome data was reannotated to identify the gene symbols based on annotation file (Homo_sapiens.GRCh38.87.chr.gtf) downloaded from Ensemble gene browser (http://www.ensembl.org/). Only the protein coding genes were selected during the annotation. Others were filtered in this step.
Identification of differentially expressed genes (DEGS)
The Location of DEGs was the first step in our research. It played a crucial role in the studying internal mechanism in HCC [12].The identification was conducted by R/edgeR, obtained from an open-source Bioinformatics project, Bioconductor (http://www.bioconductor.org/) [13, 14]. The negative binomial distributions is the key foundation of the package, also involving empirical Bayes estimation, exact tests, generalized linear models (GLM) and quasi-likelihood tests. LogFC ≥ 2.0 or logFC ≤ −2.0 associated with the P value < 0.01 were selected as the statistically significant difference.
Gene Ontology and KEGG pathway enrichment
The analysis of Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment is an essential aspect of Bioinformatics to reveal the biological functions and molecular mechanisms of DEGs [15, 16]. DAVID database (https://david.ncifcrf.gov/) is designed as a web tool which contains the relevant biological annotation. We revealed the biology function and pathways of DEGs with DAVID. In this step, the false discovery rate (FDR) < 0.1 was considered as significance for filtering the GO terms and KEGG pathways.
Protein–protein interaction (PPI) network of DEGS
Search Tool for the Retrieval of Interacting Genes (STRING) database (http://www.string-db.org/) is an important Bioinformatics tool for determining the relationship between genes [17]. We performed the PPI analysis in order to promote our understanding of undetected connection underlying the DEGs. Here, we choose only the experimentally validated PPI links with its combined score > 0.7 to enhance the reliability. Nodes with no links with others were discarded. We defined a C-score (Connection score) to measure the hub degree for every node. The genes with a high C-score had the potential to be the biomarkers. We screened the genes for their C-score ≥ 5 as the significance.
Significant biomarkers selection
The expression of significant hub genes got from PPI analysis was log2 scale. For the prognostic signature analysis, the 369 HCC samples that contained complete clinical data were assigned into groups of good or poor prognosis according to the 5 years survival (expected survival time > 5 or < 5 years). Recursive Feature Elimination (RFE) based on Random Forest (RF) classifier, a method of supervised Machine Learning, was conducted to identify the prognostic genes in survival [18, 19]. The prediction was examined by fivefolds cross-validation. We selected the best prognostic genes according to the accuracy of the RFE-RF predictor. Genes selected by RFE-RF were chosen as the candidate biomarkers. The selection was performed with R/Caret package.
Survival model
The genes selected from RFE-RF were considered as the variables for survival analysis. We separated the 369 patients with complete clinical data into training (n = 239) and testing (n = 130) datasets randomly. To better investigate the performance of these genes in predicting survival, multivariate Cox regression model was conducted in the training dataset. The coefficients were used as the weight for genes’ expression to create a risk score model. Besides, samples were divided into two groups according to the median value of risk score model. Keplan-Meire (KM) method was performed to test the prognostic performance of the model. All the analysis was used with R/survival package.
Patients’ information and tissue samples
Tissue samples were taken from patients who had undergone liver resections in the Second Hospital of Lanzhou University. All patients received liver resections from July 2012 to December 2014. None of the patients received preoperative chemotherapy and radiotherapy. All patients were followed until December 2018. Details of the clinical traits of all patients are shown in Tables 1 and 2. All groups were assessed and unidentified based on ethical criteria. The period of time between the operation and death or final result is defined as Legislation General survival (OS). Survivorship Disease (DFS) is regared as the period elapsed between performed surgery and tumor development.
Immunohistochemical staining and antibodies
Tissue samples from 95 cases of hepatocellular carcinoma were used in formalin and paraffin embedded for NUF2 and BLM immunohistochemistry. NUF2 and BLM antibodies for staining of immunochemistry were obtained from ABCAM (ab230313 and ab62206). After defrost, moisture, and embolism, samples were mixed with NUF2’s primary immunoglobulin antibodies and BLM antibodies and then incubated at night at 4 (dilution ratio 1: 1000). Finally, all sections were evaluated by comparing the staining of each sample of cancer cells from the liver and the normal sample under a microscope. The positive cell score and color intensity determine the overall score. The degree of intensity of the coloration is as bellow: 0: no coloration. 1: slightly yellowish in the background, 2: yellow and brown. 3: Brown. The positive result of the cell is as follows: 0 degree: 0–5%; 1 degree: 6–25%; 2 degree: 26–50%; 3 degree: 51–75%; 4 degree: > 75%. The overall result of immunochemistry was calculated as a positive degree of x-cell staining intensity. The overall score is divided by four levels: 0 means negative (−), 1–4 defined as positive weakness (+), 5–8 stand for positive (++) and 9–12 regared strongly positive (+++).
Statistical analysis
All data analyses were conducted with edgeR. Evaluation of patient samples were evaluated using Pierson correlation coefficients. Survival rate calculated using Cox proportional hazard model. Survival curves were calculated by the Kaplan–Meier method.
Results
Identification of DEGs
Identification of the DEGs in HCC samples demonstrated that there were 1844 up-regulated DEGS and 213 down-regulated DEGS, based on R/edgeR. The heat map of the DEGs (top 50 up-regulated and down-regulated genes according to the LogFC) is shown as an example (Fig. 1).

Heatmap of top 50 up-regulated and down-regulated genes with the highest LogFC
GO and KEGG pathway enrichment
The enriched GO terms were totally separated in three groups biological process (BP), cellular component (CC) and molecular function (MF). The result of GO enrichment showed DEGs participated in lots of significant biology processes, such as extracellular region, sequence-specific DNA binding, extracellular space and transcriptional activator activity. KEGG pathways enrichment demonstrated DEGs were mapped on the several important pathways, including cell cycle and neuroactive ligand-receptor interaction (Fig. 2).

GO terms and KEGG pathways enrichment of DEGs with FDR < 0.1. (a Biological_processes, b Cellular_component, c Molecular_function, D. KEGG pathways)
PPI analysis and biomarker selection
26 hub genes were screened from the PPI network for their hub degree ≥ 5. Among these hub genes, CDK1 showed the highest node degree, which was 22. We illustrated the Circos map of the hub genes to disclose their location on the chromosome and the links with others (Fig. 3). Besides, 5 genes including SPC25, MCM2, NUF2, AURKA and BLM were screened from the RFE-RF method. The accuracy of five candidate biomarkers for predicting prognosis reaches 0.89 (Fig. 4).

Circos Map of hub genes in PPI analysis, including 8 layers. From outside to inside: chromosome; hub genes with C-score > 10; hub genes with C-score ≥ 5; hub genes with C-score < 5; LogFC of hub genes; expression in normal tissues; expression in tumor tissues; PPI interaction links

The relationship between variables and accuracy in RFE-RF predictor, with the fivefolds cross-validation
Risk score survival model of 5 biomarkers
The risk score model was carried out by multivariate Cox regression model. The coefficients of the result were used as the weight for each gene to create a risk score model. Risk score = (0.3497 × expression level of SPC25) + (0.0995 × expression level of MCM2) + (0.0327 × expression level of NUF2) + (0.0369 × expression level of AURKA) + (-0.3185 × expression level of BLM). The risk score model was examined in the testing group and full dataset with KM curve and P value (Figs. 5 and 6). The patients with higher risk scores had the worse survival compared with lower ones (Fig. 7). Risk score had the negative correlation with overall survival (OS). The analysis suggested risk score model can be considered as an independent clinical feature for OS of the patients with HCC.

Performance of the risk score model in training (n = 239) and testing (n = 130) groups, examined by Keplan-meier method

Performance of risk score model in full dataset (n = 369), examined by Keplan-meier method

Performance validation of risk score model. a Risk score distribution, b survival time of the patients, sorted by risk score, c expression pattern of five prognostic biomarkers in 369 patients
Overexpression of NUF2 and BLM imply poor survival in patients with HCC
To ascertain the prognostic value of NUF2 and BLM, We performed a KM analysis, and all results are detailed in Fig. 8. Hyper Expression of NUF2 and BLM shown lowest OS and DFS. Next, Cox relative risk model is used to examine Whether NUF2 and BLM can become independent diagnostic influence factor for 95 patients suffering from liver cancer in our center. Results show poor prognosis in patients with high expression of NUF2 and BLM (Fig. 8). Multivariate analysis showed that the expression NUF2 and BLM (HR 2.35, 95% CI 1.06–6.11, P < 0.05) were independent predictors of the operating system. Therefore, we can think that nuf2 and BLM can provide independent prediction for liver cancer patients.

a, c NUF2 and BLM expressions are correlated with clinic-pathological features and poor prognosis. Immunohistochemical staining showed low NUF2 and BLM expressions in normal liver tissues and HCC tissues. The scales bars indicate 50 μm and 20 μm. b, d Overall survival and disease-free survival curves for HCC patient groups. All *P < 0.05, **P < 0.01
Discussion
Although many gene products affecting liver cancer have been discovered, the molecular mechanisms underlying the occurrence and development of HCC are still unclear. Thereafter, it is useful to improve the diagnosis in patients with liver cancer by detecting the vital signs that contribute to the diagnosis and treatment of liver cancer [20]. These changes can control the global regulatory mechanisms that lead to collaboration between different metabolic pathways and different signals. Therefore, the cross-interaction genes examined from these associated pathways could be the major biomarkers of HCC.
In this study, HCC RNA expression profiles were downloaded for HCC and DEGs were examined. A number of 1844 regular genes were extracted from tumor samples and 213 regular genes were obtained from subcancerous liver tissue, which were assigned to a compact PPI network. Then select pivotal genes based on RFE-RF predictor accuracy and node degree in the PPI network [21]. A number of 100 pivotal genes were detected. KEGG analysis of the enrichment pathway implied that these pivotal genes were remarkably enriched in 22 pathways including pathways in the cell cycle and interaction between neuron receptor receptors, which were Reported to be ramarkbaly related to the occurrence of liver cancer [10, 22]. We then categorized 100 survival genes through random survival forests, the most important biomarkers are SPC25, MCM2, NUF2, AURKA and BLM.
Some studies have recently shown that unregulated SPC25 is associated with the carcinogenic process and malignant patterns of certain tumors. The regulation of SPC25 is found in colorectal and gastric cancers [23]. It acts as a gene systematically linked to liver cancer, associated with early recurrences after curative resection [24]. Many clinical parameters such as advanced tumor score, advanced stage, and poor prognosis in malignancies are highly related with MCM2 [25,26,27]. In addition, their study revealed that the cytoplasmic compound MCM2-gp70 associated with protein phosphatase 2A (PP2A) interferes with the PP2A-DNA-PK reaction and promotes apoptosis caused by DNA damage by activating p53 by DNA-PK [28]. NUF2 has been discovered to participate in cancerous tumors of many types of human tumors. Previous studies suggest that depletion of NUF2 by specific siRNAs inhibit proliferation and induce apoptosis in non-small cell and ovarian cancer cells [23, 29, 30]. Similarly, a reduction in NUF2 inhibited tumor growth caused by apoptosis in human tumor cells [31]. In addition, NUF2 played a key role in pancreatic cancer profiles by regulating RNA lnc RNA 339813 [32]. It has been shown that Aurka was involved in many cancers and was aneuploidy and genetic instability [33, 34]. The main functional partner proteins include inhibitors of MYCN, NFKBa, AKT1, RALA, P53 and BRCA1 [33, 35,36,37,38,39,40]. AURKA regulates the phosphorylation of these important carcinogenic proteins leading to their respective pathways. Other evidence of PLA, FAK and Src [41]. Bloom syndrome (Bloom’s Syndrome, BLM), a member of the Recase helase family, is one of the essential vaccines required for the metabolic processes of DNA, including recombination, redundancy, and repair of DNA. DNA. It is known that the level of BLM expression is regulated differently during cell cycle stages and is expressed at high levels in cancer cells. Since BLM abnormalities are associated with genome instability, evidence accumulates in various cancers [42,43,44]. Previous studies have also revealed that the role of BLM in p53 is binding in the Chk1 pathway [45].
Conclusions
We collected a highly reliable database of hepatocellular carcinoma and used these datasets to build a survival prediction model based on the above 5 genes through multi-variable Cox regression. This risk score predicted patients at high risk of mortality independently. Immunohistochemical experiments were performed, and the results shown that NUF2 may play an pivotal role in promoting the occurrence and development of liver cancer, but the mechanism needs more research to demonstrate and and this is what we are doing. Our current work aims to provide the fresh methods for the clinical application of gene expression profiling in HCC, Especially in the future, this method will be applied to individualized prediction of disease and precision medicine, But the reliability and accuracy of this risk assessment must be verified through more forward-looking studies.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the TCGA repository. http://www.tcga.org
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- TCGA:
-
The Cancer Genome Atlas
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein and protein interaction
- RFE:
-
Recursive feature elimination
- RF:
-
Random forest
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45(4):529–38.
Marrero JA, Kudo M, Bronowicki JP. The challenge of prognosis and staging for hepatocellular carcinoma. Oncologist. 2010;15(Suppl 4):23–33.
Fantini M, Benvenuto M, Masuelli L, Frajese GV, Tresoldi I, Modesti A, Bei R. In vitro and in vivo antitumoral effects of combinations of polyphenols, or polyphenols and anticancer drugs: perspectives on cancer treatment. Int J Mol Sci. 2015;16(5):9236–82.
Hai H, Tamori A, Kawada N. Role of hepatitis B virus DNA integration in human hepatocarcinogenesis. World J Gastroenterol. 2014;20(20):6236–43.
Takai A, Dang HT, Wang XW. Identification of drivers from cancer genome diversity in hepatocellular carcinoma. Int J Mol Sci. 2014;15(6):11142–60.
Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12(3):170–80.
Astolfi A, Landuzzi L, Nicoletti G, De Giovanni C, Croci S, Palladini A, Ferrini S, Iezzi M, Musiani P, Cavallo F, et al. Gene expression analysis of immune-mediated arrest of tumorigenesis in a transgenic mouse model of HER-2/neu-positive basal-like mammary carcinoma. Am J Pathol. 2005;166(4):1205–16.
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–10.
Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS 3rd, Johnson BE, Skolnick MH. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264(5157):436–40.
Yun C, Lee S. The roles of autophagy in cancer. Int J Mol Sci. 2018;19(11):3466–3484. https://doi.org/10.3390/ijms19113466
Rosok O, Sioud M. Discovery of differentially expressed genes: technical considerations. Methods Mol Biol. 2007;360:115–29.
Law CW, Alhamdoosh M, Su S, Dong X, Tian L, Smyth GK, Ritchie ME. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research. 2016. https://doi.org/10.12688/f1000research.9005.3.
Reimers M, Carey VJ. Bioconductor: an open source framework for bioinformatics and computational biology. Methods Enzymol. 2006;411:119–34.
Du J, Yuan Z, Ma Z, Song J, Xie X, Chen Y. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol BioSyst. 2014;10(9):2441–7.
Gene Ontology C. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006;34(Database issue):D322–6.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Le TT, Simmons WK, Misaki M, Bodurka J, White BC, Savitz J, McKinney BA. Differential privacy-based evaporative cooling feature selection and classification with relief-F and random forests. Bioinformatics. 2017;33(18):2906–13.
Ravishankar H, Madhavan R, Mullick R, Shetty T, Marinelli L, Joel SE. Recursive feature elimination for biomarker discovery in resting-state functional connectivity. Conf Proc. 2016;2016:4071–4.
Zinkin NT, Grall F, Bhaskar K, Otu HH, Spentzos D, Kalmowitz B, Wells M, Guerrero M, Asara JM, Libermann TA, et al. Serum proteomics and biomarkers in hepatocellular carcinoma and chronic liver disease. Clin Cancer Res. 2008;14(2):470–7.
Heo M, Maslov S, Shakhnovich E. Topology of protein interaction network shapes protein abundances and strengths of their functional and nonspecific interactions. Proc Natl Acad Sci USA. 2011;108(10):4258–63.
Fabregat I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J Gastroenterol. 2009;15(5):513–20.
Kaneko N, Miura K, Gu Z, Karasawa H, Ohnuma S, Sasaki H, Tsukamoto N, Yokoyama S, Yamamura A, Nagase H, et al. siRNA-mediated knockdown against CDCA1 and KNTC2, both frequently overexpressed in colorectal and gastric cancers, suppresses cell proliferation and induces apoptosis. Biochem Biophys Res Commun. 2009;390(4):1235–40.
Chen J, Rajasekaran M, Xia H, Zhang X, Kong SN, Sekar K, Seshachalam VP, Deivasigamani A, Goh BK, Ooi LL, et al. The microtubule-associated protein PRC1 promotes early recurrence of hepatocellular carcinoma in association with the Wnt/beta-catenin signalling pathway. Gut. 2016;65(9):1522–34.
Davies RJ, Freeman A, Morris LS, Bingham S, Dilworth S, Scott I, Laskey RA, Miller R, Coleman N. Analysis of minichromosome maintenance proteins as a novel method for detection of colorectal cancer in stool. Lancet. 2002;359(9321):1917–9.
Dudderidge TJ, Stoeber K, Loddo M, Atkinson G, Fanshawe T, Griffiths DF, Williams GH. Mcm2, Geminin, and KI67 define proliferative state and are prognostic markers in renal cell carcinoma. Clin Cancer Res. 2005;11(7):2510–7.
Majid S, Dar AA, Saini S, Chen Y, Shahryari V, Liu J, Zaman MS, Hirata H, Yamamura S, Ueno K, et al. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res. 2010;70(7):2809–18.
Abe S, Kurata M, Suzuki S, Yamamoto K, Aisaki K, Kanno J, Kitagawa M. Minichromosome maintenance 2 bound with retroviral Gp70 is localized to cytoplasm and enhances DNA-damage-induced apoptosis. PLoS ONE. 2012;7(6):e40129.
Hayama S, Daigo Y, Kato T, Ishikawa N, Yamabuki T, Miyamoto M, Ito T, Tsuchiya E, Kondo S, Nakamura Y. Activation of CDCA1-KNTC2, members of centromere protein complex, involved in pulmonary carcinogenesis. Cancer Res. 2006;66(21):10339–48.
Sethi G, Pathak HB, Zhang H, Zhou Y, Einarson MB, Vathipadiekal V, Gunewardena S, Birrer MJ, Godwin AK. An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. PLoS ONE. 2012;7(10):e47086.
Huang SK, Qian JX, Yuan BQ, Lin YY, Ye ZX, Huang SS. SiRNA-mediated knockdown against NUF2 suppresses tumor growth and induces cell apoptosis in human glioma cells. Cell Mol Biol (Noisy-le-grand). 2014;60(4):30–6.
Hu P, Shangguan J, Zhang L. Downregulation of NUF2 inhibits tumor growth and induces apoptosis by regulating lncRNA AF339813. Int J Clin Exp Pathol. 2015;8(3):2638–48.
Karthigeyan D, Prasad SB, Shandilya J, Agrawal S, Kundu TK. Biology of Aurora A kinase: implications in cancer manifestation and therapy. Med Res Rev. 2011;31(5):757–93.
Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70(4):661–87.
Blanco I, Kuchenbaecker K, Cuadras D, Wang X, Barrowdale D, de Garibay GR, Librado P, Sanchez-Gracia A, Rozas J, Bonifaci N, et al. Assessing associations between the AURKA-HMMR-TPX2-TUBG1 functional module and breast cancer risk in BRCA1/2 mutation carriers. PLoS ONE. 2015;10(4):e0120020.
Briassouli P, Chan F, Savage K, Reis-Filho JS, Linardopoulos S. Aurora-A regulation of nuclear factor-kappaB signaling by phosphorylation of IkappaBalpha. Cancer Res. 2007;67(4):1689–95.
den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116(9):1498–505.
Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, Simonds EF, Seeger R, Matthay KK, Hertz NT, et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell. 2014;26(3):414–27.
Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD, Counter CM. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010;30(2):508–23.
Wang Y, Sun H, Wang Z, Liu M, Qi Z, Meng J, Sun J, Yang G. Aurora-A: a potential DNA repair modulator. Tumour Biol. 2014;35(4):2831–6.
Mahankali M, Henkels KM, Speranza F, Gomez-Cambronero J. A non-mitotic role for Aurora kinase A as a direct activator of cell migration upon interaction with PLD, FAK and Src. J Cell Sci. 2015;128(3):516–26.
Davari P, Hebert JL, Albertson DG, Huey B, Roy R, Mancianti ML, Horvai AE, McDaniel LD, Schultz RA, Epstein EH Jr. Loss of Blm enhances basal cell carcinoma and rhabdomyosarcoma tumorigenesis in Ptch1 ± mice. Carcinogenesis. 2010;31(6):968–73.
de Voer RM, Hahn MM, Mensenkamp AR, Hoischen A, Gilissen C, Henkes A, Spruijt L, van Zelst-Stams WA, Kets CM, Verwiel ET, et al. Deleterious germline blm mutations and the risk for early-onset colorectal cancer. Sci Rep. 2015;5:14060.
Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, Thorne H, Barnes DR, Li J, kConFab, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8(9):e1002894.
Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J, et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol. 2004;166(6):801–13.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (Grant No. 31770537). Lanzhou Science and Technology Bureau Project (2017-RC-64). Gansu Administration of Traditional Chinese Medicine Project (GZK-2019-47). Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (CY2017-QN20)
Author information
Authors and Affiliations
Contributions
LG, YD and JM performed the majority of experiments; LG and YL Performed bioinformatics analysis, JZ and HZ provided vital reagents and analytical tools and were also involved in editing the manuscript; ZY and JG co-ordinated and provided the collection of all the human material; Lingyun Guo and Zhenjiang Wang designed the study and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval for the research study was obtained from the Lanzhou University Second Hosptial Ethics Board (project approval number 2019-026).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, L., Wang, Z., Du, Y. et al. Random-forest algorithm based biomarkers in predicting prognosis in the patients with hepatocellular carcinoma. Cancer Cell Int 20, 251 (2020). https://doi.org/10.1186/s12935-020-01274-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01274-z