
Abstract
Background
The chromosome 3q29 microdeletion syndrome is characterized by a clinical phenotype that includes behavioral features consistent with autism and attention deficit hyperactivity disorder, mild to moderate developmental delay, language-based learning disabilities, and/or dysmorphic features. In addition, recent data suggest that adults with chromosome 3q29 microdeletions have a significantly increased risk for psychosis and neuropsychiatric phenotypes.
Case presentation
We report a 3-year-old male with global developmental delay, anemia, and mild dysmorphic facial features. Clinical chromosomal microarray (CMA) testing of the proband detected a heterozygous 1.21 Mb deletion at chromosome 3q29, consistent with a diagnosis of the 3q29 microdeletion syndrome. Interestingly, subsequent parental testing determined that the pathogenic deletion was inherited from his otherwise healthy mother who had a history of learning disabilities. The chromosome 3q29 microdeletion was not detected in the healthy older sibling of the proband by CMA testing, nor was it prenatally detected in a subsequent maternal pregnancy.
Conclusion
Our report highlights the 3q29 microdeletion syndrome as an illustrative example of the importance of a molecular diagnosis for families that harbor pathogenic copy number aberrations with variable expressivity, in particular those that also impart an increased risk for adult onset neuropsychiatric phenotypes.
Similar content being viewed by others


Background
Chromosomal microarray (CMA) testing offers a powerful approach to detect submicroscopic copy number changes in the human genome [1, 2], which has led to the discovery of several microdeletion and microduplication syndromes [3, 4]. Moreover, clinically-relevant intragenic copy number changes are now also being discovered by large-scale genome sequencing efforts [5,6,7]. Importantly, some of these syndromes have incomplete penetrance and/or variable expressivity, which often result in nonspecific neurodevelopmental clinical features and challenging clinical diagnoses. The chromosome 3q29 microdeletion syndrome is characterized by a clinical phenotype that can include behavioral features consistent with autism and attention deficit hyperactivity disorder, mild to moderate developmental delay, and language-based learning disabilities [8]. A subset of cases also present with microcephaly and mild dysmorphic features (e.g., elongated face, long fingers, and joint laxity) [9, 10].
The majority of 3q29 microdeletion syndrome cases occur de novo [11,12,13,14,15], which are most likely derived by the regional low copy repeats that act as substrates for non-allelic homologous recombination [8]. The recurrent 3q29 microdeletion spans ~ 1.6 Mb with molecular boundaries encompassing the PAK2 and DLG1 candidate genes [8, 16]. Of note, inheritance from a mildly affected parent has also been reported among several independent families, including this index case (Table 1) [11, 14, 17,18,19,20]. This variable expression suggests that the incidence of the 3q29 microdeletion syndrome may potentially be higher than previously estimated (1 in 30,000-40,000) [21], as the constellation of features among certain families may not be clinically overt. Importantly, the 3q29 microdeletion is also one of the largest known risk factors for schizophrenia (~ 40 fold), surpassing even the recurrent 22q11.2 microdeletion syndrome [16]. As such, accurate detection and molecular diagnosis of the 3q29 microdeletion syndrome is critical for proper genetic counseling and when providing neuropsychiatric risk information to affected family members. Herein, we report a child with global developmental delay and a chromosome 3q29 microdeletion, which subsequently was found to be inherited from his otherwise healthy mother who had a history of learning disabilities. This informative case underscores the utility of CMA testing for non-specific neurodevelopmental pediatric indications and highlights the 3q29 microdeletion syndrome as an illustrative example of the importance of a molecular diagnosis for pathogenic copy number aberrations with variable expressivity.
Case presentation
The 3-year-old male proband was referred to Clinical Genetics for evaluation of a history of developmental delay. He was the second child of non-consanguineous parents of Mexican descent, and his pedigree was remarkable for a mother and maternal male first cousin who required special education mainly for learning disabilities. The prenatal history included a maternal chlamydia infection at 3 months gestational age (treated with antibiotics) and intrauterine growth restriction. He was born vaginally at 37 weeks gestational age, weighing 1.98 kg, and spent a total of 23 days in the NICU requiring nasogastric feeds and phototherapy. He walked and began to speak at 14 months of age and was subsequently diagnosed with global developmental delay by a behavioral pediatrician. He began early intervention services at the age of 2 years, receiving occupational, physical and speech therapy. At 3 years of age he was placed in special education classes getting speech and occupational therapies. At the time of his initial genetics evaluation, the patient could understand directions and point to body parts. He did not know the alphabet, colors, or numbers. In addition to these developmental delays, the proband has a history of anemia that is followed by his pediatrician. On physical exam, the patient was found to have a low anterior hair line. His palpebral fissures were slightly downward slanting, and infraorbital puffiness was noted. He has overfolded helices bilaterally, a broad nasal bridge, a wide grin and thin upper lip, which generally resembled the facial features of his mother (Fig. 1).

Facial features of the proband and his affected mother. Note the similar broad nasal bridge seen in both the parent and proband (a). A closer look at the proband reveals periorbital puffiness and slightly downward slanting palpebral fissures (b). Profile views of the proband and his mother demonstrate similar ears with an overfolded helix (c-d)
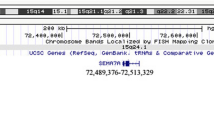
Clinical Fragile X testing on DNA isolated from the proband was normal with 29 CGG repeats; however, CMA testing using the SurePrint G3 ISCA CGH + SNP 4 × 180 microarray (Agilent Technologies, Santa Clara, CA) [22, 23] detected a heterozygous 1.21 Mb deletion of chromosome 3q29 (reported as arr[GRCh37] 3q29(195804728_197016624)× 1) (Fig. 2). This microdeletion is smaller than the 1.6 Mb recurrent 3q29 microdeletion defined above, but still nested within its segmental duplication boundaries (Fig. 2). It included 28 genes and transcripts, and had only minimal overlap with copy number variants (CNVs) reported among healthy individuals in the Database of Genomic Variants (DGV; http://dgv.tcag.ca) [24]. The interstitial chromosome 3q29 microdeletion was confirmed using the higher resolution CytoScan® HD platform (Affymetrix, Santa Clara, CA) and metaphase fluorescent in situ hybridization (FISH) using subtelomeric DNA probes (Fig. 2). Notably, familial CMA testing determined that the pathogenic chromosome 3q29 microdeletion was maternally inherited and not present in the healthy older sibling (Fig. 2), nor was it prenatally detected in a subsequent maternal pregnancy.

Chromosome view showing the subtelomeric 3q29 microdeletion region. Each red and blue dot represents an oligonucleotide probe along the length of chromosome 3 with its cytobands shown on the x-axis and the log2 ratio plotted on the y-axis. The dotted lines provide a focused view of a portion the 3q29 cytoband, annotations of UCSC genes (hg19) (http://genome.ucsc.edu/), user-defined tracks indicating the recurrent 1.6 Mb deletion interval [(reported in [10]], and the 1.21 Mb deletion reported in our index patient. Immediately below, the presence of copy number variants (CNVs) annotated in the Database of Genomic Variants (DGV; blue: gain; red: loss) and region-specific segmental duplications (SDs) are shown in context (a). Zoomed-in views of chromosomal microarray (CMA) plots (X axis: hg19 genomic coordinates; Y axis: mean log2 ratio; Agilent Technologies) depicting heterozygous copy number loss in the proband (top panel). Refinement of the deletion with a higher resolution CMA platform (Affymetrix; bottom panel arr[GRCh37]3q29(195806608_197029439)× 1) (b). CMA plots of the mother detailing the same heterozygous deletion as in the proband, whereas the sibling did not inherit the pathogenic chromosome 3q29 deletion (c). Metaphase and interphase FISH specific to the subtelomeric region of chromosome 3q (D3S4560 Abbott Molecular TelVysion, red signal), and an internal control probe specific to the subtelomeric region of chromosome 3p (D3S4559 Abbott Molecular TelVysion, green signal) in the proband. A red signal is absent at 3q29 on one of the chromosome 3 homologs (arrow), which confirms the heterozygous deletion identified by CMA testing (d)
Discussion and conclusions
The significance of structural variation in human disease and phenotypic diversity has increasingly become recognized, and several genomic studies have generated catalogs of CNVs to facilitate a better understanding of their clinical relevance [3, 25, 26]. It is estimated that up to 60% of the human genome may contain structural variants in the form of CNVs in the general population, which typically range in size from 100 bp to 50 kb [27], and clinical interpretation of these aberrations when identified by CMA testing is enabled by medical genetics practice guidelines [28]. Larger copy number aberrations are more likely to be pathogenic, as are de novo variants; however, large-scale genomic studies on clinically relevant sequence variants and CNVs are increasingly underscoring the importance and interpretation challenges of incomplete penetrance and variable expressivity [3, 29,30,31]. We report a case of the 3q29 microdeletion syndrome in a 3-year-old male with developmental delay, which was found to be inherited from an otherwise healthy parent with a history of learning disabilities (Table 1). This educational case is an illustrative example of the importance of a molecular diagnosis for children with non-specific neurodevelopmental indications, particularly for families that harbor pathogenic copy number aberrations with variable expressivity.
As indicated above, the recurrent 3q29 microdeletion syndrome has a heterogeneous clinical phenotype, and is enriched among young adults with psychiatric disorders and children with developmental delay [3, 32]. In particular, mild to moderate cognitive deficits, speech delay, low birth weight, a high nasal root, and ocular abnormalities are common findings (Table 1). Other less frequent features that can present with greater variability include facial asymmetry, frontal bossing, large protuberant ears, upslanting palpebral fissures, broad nostrils, and cleft lip/palate [11,12,13,14,15].
The 1.21 Mb deletion identified in our proband is nested within the recurrent ~ 1.6 Mb deletion and is one of the smallest 3q29 microdeletions reported to date using a high-resolution CMA platform; however, it directly overlaps with previously described pathogenic deletions at this locus [21]. Additional 3q29 deletions nested within the recurrent microdeletion have also been described, either with a BAC or low resolution oligonucleotide microarray platform [12, 13, 17, 33]. A common pattern of moderate learning problems, speech delay, psychiatric disorder, and autistic features constitutes the primary phenotypic overlap in this cohort of smaller 3q29 deletions. A comparison of the genomic intervals between the 1.21 Mb deletion in our proband, the previously reported smaller deletions [12, 13, 17, 33], and the more common recurrent 1.6 Mb deletions indicates that all include the PAK2 candidate gene. The centromeric region of all deletions share the TFRC gene, which is also disrupted by the deletion in our proband and focally deleted in the recurrent deletion (Fig. 2). The telomeric region commonly includes DLG1; however, the recurrent larger deletions extend past DLG1 and into BHD1 (Fig. 2). Haploinsufficiency of these genes, in particular DLG1, has been implicated in some forms of schizophrenia and the behavioral characteristics of the 3q29 microdeletion syndrome [16, 17, 34].
In conclusion, our case presents a set of phenotypic features that add to the heterogeneous expression of the 3q29 microdeletion syndrome. In particular, the finding of anemia in our proband is uncommon among reported 3q29 microdeletion syndrome cohorts; however, given that the co-occurrence of anemia may be coincidental, additional patients are needed to confirm whether this is a rare feature of the 3q29 microdeletion. The familial inheritance of this pathogenic deletion and the phenotype of the transmitting parent is summarized with other reported familial 3q29 microdeletion syndrome cases in Table 1. Consistent among these families is a milder phenotype in the transmitting parent, often restricted to mild dysmorphic features and/or a history of learning/language delays. As the landscape of genomic variation in different populations continues to be defined, the importance of incomplete penetrance and variable expressivity are increasingly emerging. As such, the familial inheritance of this pathogenic deletion highlights both the clinical relevance of variable expressivity in genetic disease and the importance of clinician awareness, particularly for inherited aberrations that also impart an increased risk for adult onset neuropsychiatric phenotypes.
Abbreviations
- CMA:
-
chromosomal microarray
- CNV:
-
copy number variant
- DGV:
-
Database of Genomic Variants
- FISH:
-
fluorescence in situ hybridization
References
Kearney HM, South ST, Wolff DJ, Lamb A, Hamosh A, Rao KW, et al. American College of Medical Genetics recommendations for the design and performance expectations for clinical genomic copy number microarrays intended for use in the postnatal setting for detection of constitutional abnormalities. Genet Med. 2011;13(7):676–9.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–64.
Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46(10):1063–71.
Edelmann L, Hirschhorn K. Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies. Ann N Y Acad Sci. 2009;1151:157–66.
Salpietro V, Manole A, Efthymiou S, Houlden H. A review of copy number variants in inherited neuropathies. Current genomics. 2018;19(6):412–9.
Pfundt R, Del Rosario M, Vissers LE, Kwint MP, Janssen IM, De Leeuw N, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genetics in Medicine. 2017;19(6):667.
Zare F, Dow M, Monteleone N, Hosny A, S N. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinformat. 2017;18(1):286.
Willatt L, Cox J, Barber J, Cabanas ED, Collins A, Donnai D, et al. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am J Hum Genet. 2005;77(1):154–60.
Cobb W, Anderson A, Turner C, Hoffman RD, Schonberg S, Levin SW. 1.3 Mb de novo deletion in chromosome band 3q29 associated with normal intelligence in a child. Eur J Med Genet. 2010;53(6):415–8.
Quintero-Rivera F, Sharifi-Hannauer P, Martinez-Agosto JA. Autistic and psychiatric findings associated with the 3q29 microdeletion syndrome: case report and review. Am J Med Genet A. 2010;152A(10):2459–67.
Clayton-Smith J, Giblin C, Smith RA, Dunn C, Willatt L. Familial 3q29 microdeletion syndrome providing further evidence of involvement of the 3q29 region in bipolar disorder. Clin Dysmorphol. 2010;19(3):128–32.
Cox DM, Butler MG. A clinical case report and literature review of the 3q29 microdeletion syndrome. Clin Dysmorphol. 2015;24(3):89–94.
Dasouki MJ, Lushington GH, Hovanes K, Casey J, Gorre M. The 3q29 microdeletion syndrome: report of three new unrelated patients and in silico "RNA binding" analysis of the 3q29 region. Am J Med Genet A. 2011;155A(7):1654–60.
Li F, Lisi EC, Wohler ES, Hamosh A, Batista DA. 3q29 interstitial microdeletion syndrome: an inherited case associated with cardiac defect and normal cognition. Eur J Med Genet. 2009;52(5):349–52.
Wang JC, Naik H, Khan A, Nowaczyk MJ. An uncommon 3.4-Mb interstitial deletion at 3q29. Clin Dysmorphol. 2010;19(3):133–6.
Mulle JG. The 3q29 deletion confers >40-fold increase in risk for schizophrenia. Mol Psychiatry. 2015;20(9):1028–9.
Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, Madan-Khetarpal S, et al. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol Cytogenet. 2008;1:8.
Monfort S, Rosello M, Orellana C, Oltra S, Blesa D, Kok K, et al. Detection of known and novel genomic rearrangements by array based comparative genomic hybridisation: deletion of ZNF533 and duplication of CHARGE syndrome genes. J Med Genet. 2008;45(7):432–7.
Digilio MC, Bernardini L, Mingarelli R, Capolino R, Capalbo A, Giuffrida MG, et al. 3q29 Microdeletion: a mental retardation disorder unassociated with a recognizable phenotype in two mother–daughter pairs. AM J Med Genet Part A. 2009;149(8):1777–81.
Petrin AL, Daack-Hirsch S, L'heureux J, Murray JC. A case of 3q29 microdeletion syndrome involving oral cleft inherited from a nonaffected mosaic parent: molecular analysis and ethical implications. Cleft Palate Craniofac J. 2011;48(2):222–30.
Glassford MR, Rosenfeld JA, Freedman AA, Zwick ME, Mulle JG. Unique rare chromosome disorder support G. Novel features of 3q29 deletion syndrome: results from the 3q29 registry. Am J Med Genet A. 2016;170A(4):999–1006.
Scott SA, Cohen N, Brandt T, Toruner G, Desnick RJ, Edelmann L. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet Med. 2010;12(2):85–92.
Reiner J, Karger L, Cohen N, Mehta L, Edelmann L, Scott SA. Chromosomal microarray detection of constitutional copy number variation using saliva DNA. J Mol Diagn. 2017;19(3):397–403.
MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(Database issue):D986–92.
Johansson AC, Feuk L. Characterization of copy number-stable regions in the human genome. Hum Mutat. 2011;32(8):947–55.
Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81.
Escaramis G, Docampo E, Rabionet R. A decade of structural variants: description, history and methods to detect structural variation. Brief Funct Genomics. 2015;14(5):305–14.
South ST, Lee C, Lamb AN, Higgins AW, Kearney HM, Working Group for the American College of medical G, et al. ACMG standards and guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet Med. 2013;15(11):901–9.
Van Driest SL, Wells QS, Stallings S, Bush WS, Gordon A, Nickerson DA, et al. Association of Arrhythmia-Related Genetic Variants with Phenotypes Documented in electronic medical records. Jama. 2016;315(1):47–57.
Nowakowska B. Clinical interpretation of copy number variants in the human genome. J Appl Genet. 2017;58(4):449–57.
Brabbing-Goldstein D, Reches A, Svirsky R, Bar-Shira A, Yaron Y. Dilemmas in genetic counseling for low-penetrance neuro-susceptibility loci detected on prenatal chromosomal microarray analysis. Am J Obstet Gynecol. 2018;218(2):247. e1-. e12.
Kirov G, Rees E, Walters JT, Escott-Price V, Georgieva L, Richards AL, et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry. 2014;75(5):378–85.
Mulle JG, Dodd AF, McGrath JA, Wolyniec PS, Mitchell AA, Shetty AC, et al. Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet. 2010;87(2):229–36.
Carroll LS, Williams HJ, Walters J, Kirov G, O'Donovan MC, Owen MJ. Mutation screening of the 3q29 microdeletion syndrome candidate genes DLG1 and PAK2 in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(7):844–9.
Acknowledgements
The authors would like to thank the proband and family for supporting publication of this case report.
Funding
None.
Availability of data and materials
The datasets generated during and/or analysed during the current study are not publicly available due to these data being derived from clinical genetic testing; however, deidentified data may be available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
WAK performed, analyzed, and interpreted the CMA and FISH testing; drafted and revised the manuscript. NC analyzed and interpreted the data and revised the manuscript. SAS interpreted the data, drafted and revised the manuscript, and oversaw the case report. EP performed the clinical evaluations, interpreted the data, drafted and revised the manuscript, and oversaw the case report. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The family provided written informed consent for the clinical genetic testing performed in this study through the Pediatric Genetics Clinic at Jacobi Medical Center.
Consent for publication
Informed written consent was obtained for publication (including images) from all family members involved in this study. A copy of the written consent is available for review by the editor of this Journal.
Competing interests
WAK and SAS are paid employees of Mount Sinai Genomics Inc. (DBA Sema4). All authors declare no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Khan, W.A., Cohen, N., Scott, S.A. et al. Familial inheritance of the 3q29 microdeletion syndrome: case report and review. BMC Med Genomics 12, 51 (2019). https://doi.org/10.1186/s12920-019-0497-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-019-0497-4