
Abstract
Background
Staphylococcus aureus is an important zoonotic pathogen which not only causes significant economic loss in livestock production but also poses a potential threat to public health. Compared with bovine and swine, the information on the colonization of S. aureus in goats is very limited. To understand the prevalence and characteristics of S. aureus in goats, we used the nasal swabs collected from apparently healthy goats to isolate S. aureus, and tested their antimicrobial susceptibility, virulence gene carrying levels, and multilocus sequence typing (MLST).
Results
In 74 nasal swabs of apparently healthy goats, 32 (43.24%) S. aureus strains were isolated and identified, most of which were susceptible to many antibiotics, except for trimethoprim, furazolidone, amoxicillin, lincomycin and roxithromycin, and the resistance incidence of which were 50%, 40.63%, 37.5%, 28.13%, and 21.88% respectively. All the isolates were methicillin-susceptible S. aureus (MSSA) and mecA-negative. Enterotoxin genes were found in 53.13% of the strains. Of which, sej was the most prevalent (21.88%), followed by seb, sec, and see with the same level (18.75%). The most prevalent combination were seb + see and seb + tst. None of the S. aureus isolates harbored sea, sed, seh, eta and etb. Multilocus sequence typing (MLST) revealed 6 new alleles (aroe-552, aroe-553, glpf-500, pta-440, yqil-482 and yqil-496) and 5 new sequence types (STs) (3431,3440,3444,3445 and 3461). Using eBURST, the 5 STs were assigned to clonal complex 522 (CC522) and a further CC with no predicted ancestor. Phylogenetic analysis of seven concatenated MLST alleles revealed that the 5 STs were grouped into cluster I composed of S. aureus mainly from goats and sheep.
Conclusion
We provide the data for prevalence of S. aureus in goats in Chongqing municipality and their characterization which will help in tracking evolution of epidemic strains and their control methods.
Similar content being viewed by others

Background
Staphylococcus aureus is an important opportunistic pathogen and the cause of infection among human, domestic and wild animals [1,2,3]. Due to its broad spectrum of inherent virulence factors, the infection of S. aureus usually plays an important role for the causing of abscesses, mastitis, pneumonia and meningitis in mammals [4,5,6]. The invasion of S. aureus in domestic animals not only causes significant economic loss in livestock production but also poses a potential threat to public health since these animals can act as the reservoir of methicillin-resistant S. aureus (MRSA) [7, 8]. The livestock-associated methicillin-resistant S. aureus (LA-MRSA) represented by clonal complex 398 (CC 398) have been shown to be able to colonize and cause serious infections in people having close contact with animals such as veterinarians, farmers and their family members [9,10,11]. In addition, both handling and consumption of products of these animals colonized by MRSA may provide a potential transmission to humans [12]. Thus, it is of importance to understand the prevalence and characterization of S. aureus colonizing the livestock.
In contrast to the studies of S. aureus infections in bovine and human, less is known on these bacteria in goats or their relevant products. In 2008, Chongqing municipality was incorporated into the national "Advantage of agricultural products regional planning" in China and recognized as the key areas for beef cattle and goats breeding. The number of live sheep and goats in Chongqing was about 2.26 million and 2.74 million slaughtered was provided by the end of 2015 (Chongqing Statistical Yearbook, 2016). However, there is no investigation on the incidences of colonization in goats by S. aureus in Chongqing. To understand the prevalence and characteristics of S. aureus in goats, we used the nasal swabs from apparently healthy goats to isolate S. aureus, tested their antimicrobial susceptibility and virulence gene carrying levels, and defined multilocus sequence typing (MLST).
Methods
Sample collection and S. aureus isolation
The nasal swabs were collected from 74 apparently healthy goats from 10 herds in 4 counties (Rongchang, Jiangjing, Zhongxian and Dazu) of Chongqing municipality, transported to the laboratory and stored at 4 °C prior to isolation. The bacteria were enriched in a common broth at 37 °C for 18 h, and then inoculated on 7.5% NaCl agar plates for cultivation at 37 °C for 24 h. The colonies suspected to be S. aureus were identified by Gram staining, colony morphology, and coagulase testing. All the presumptive S. aureus colonies were then subcultured and confirmed by the PCR amplification of nuc (a thermonuclease gene characteristic of S. aureus) [13] (Table 1) with the following conditions: denaturation for 4 min at 94 °C, followed by 30 cycles for 1 min at 94 °C, 30 s at 58 °C and 90 s at 72 °C, and a final extension at 72 °C for 3.5 min. S. sciuri was used as nuc-negative control strain.
Antimicrobial resistance profile
The S. aureus isolates were used for antimicrobial susceptibility test using disk diffusion method according to Clinical and Laboratory Standards Institute (CLSI) guidelines [14]. Following antibiotics with stated concentrations (μg/disc) were used: cefotaxime (30), ceftriaxone (30), streptomycin (10),kanamycin (30), gentamicin (10),tetracycline (30), chloramphenicol (30),lincomycin (2), macrodantin (300),furazolidone (300), vancomycin (30), norfloxacin (10),minocyline (30), amoxicillin (10), cefoxitin (30),clarithromycin (15), levofloxacin (5),roxithromycin (15),trimethoprim (5) and cefepime (30). The presence of mecA (staphylococci methicillin resistance gene) [15] (Table 1) was analyzed by PCR method under the same amplification conditions as stated earlier for nuc, ATCC 43300 (mecA positive) and ATCC 29213 (mecA negative) were used as control strains.
Detection of virulence genes
DNA extraction was done using a commercial DNA extraction kit (Dalian TaKaRa Biotechnology Co.,Ltd.) following the manufacturer’s instructions. Thirteen S. aureus virulence genes including enterotoxins (SEs), sea, seb, sec, sed, see, seg, seh, sei, sej [16,17,18]; cytotoxin pvl [19]; exfoliative toxins (ETs), eta, etb [20]; and a toxic shock syndrome toxin 1(tst) [16] (Table 1) were selected for PCR with the following amplification conditions: initial denaturation for 5 min at 94 °C, followed by 30 cycles of 94 °C for 1 min, 50 °C for 1 min (55 °C for seg and pvl), and 72 °C for 1 min, with a final elongation of 72 °C for 10 min. S. aureus reference strains including ATCC 25923 (pvl positive) and ATCC 29213 (sea, sec, see and tst positive).
Multilocus sequence typing (MLST) of S. aureus
A random selection of 13 S. aureus isolates were analyzed by MLST. The 7 housekeeping genes, carbamate kinase (arcc), shikimate dehydrogenase (aroe), glycerol kinase (glpf), guanylate kinase (gmk), phosphate acetyltransferase (pta), triosephosphate isomerase (tpi), and acetyl coenzyme A acetyltransferase (yqil) (http://saureus.mlst.net/misc/info.asp), were amplified by PCR method consisting of initial denaturation at 95 °C for 5 min, followed by a total of 35 cycles of 30 s denaturation at 94 °C, 30 s annealing at 55 °C, and 1 min elongation at 72 °C, with a final elongation step of 10 min at 72 °C. PCR products were sequenced both in forward and reverse directions by Shanghai Invitrogen Biotechnology Co., Ltd. The sequences for each locus were compared to the allele sequences through the MLST website (http://www.mlst.net). Isolates were defined by their alleles at the seven loci (allelic profile), and each allelic profile was submitted to the database to obtain a sequence type (ST) number.
Clonal complexes (CCs) clustering and phylogenetic analysis of S. aureus STs
The clustering of STs was performed using eBURST algorithm (http://eburst.mlst.net/) [21] by comparing the present dataset to that of the MLST database of 4066 STs by the end of May 2017, and ran with the default settings. Clonal complexes (CCs) were composed of STs that shared at least six alleles in common and a predicted ancestral ST and its associated single locus variants (SLVs: the variants differ at one of the seven MLST alleles from the ancestor) and double locus variants (DLVs: the variants differ at two of the seven MLST alleles from the ancestor) [3]. Along with the STs obtained in this research, 23 STs from human (ST7, 12,13, 15, 27, 88, 101 and 240), goats and sheep (ST130, 133, 398, 425, 522, 1595, 1740, 1742, 1743, 1758, 1780, 1781, 2011, 2305, 2328) [2, 9, 22, 23] were collected for phylogenetic analyses. The concatenated sequences of all seven MLST alleles for each ST were aligned using the CLUSTAL X program with default parameters followed by manual inspection. MEGA 4.0 was used to construct neighbour-joining trees [24]. Bootstrapping was performed with 1000 replicates.
Results
Isolation and identification of S. aureus
Of 74 nasal swabs of goats collected from 10 herds, 32 (43.24%) were identified as S. aureus through nuc amplification (Fig. 1), and all the S. aureus isolates were found to be coagulase positive.

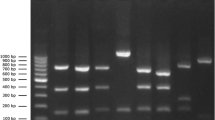
Gel electrophoresis of amplified nuc in S. aureus isolated from goats in Chongqing. M: DL-2000 DNA marker; 1: Staphylococcus sciuri; 2–5: the nuc gene of some S. aureus isolates by amplified at 297 bp
Antimicrobial resistance profile
The antimicrobial susceptibility test showed most of the S. aureus isolates susceptible to a majority of antibiotics and the resistance rates below 20%, with the exceptions of following: trimethoprim (50% resistant), furazolidone (40.63%), amoxicillin (37.5%), lincomycin (28.13%), and roxithromycin (21.88%) (Table 2). All the isolates (n = 32) were methicillin-susceptible S. aureus (MSSA) and cefoxitin-susceptible.
Distribution of virulence genes among S. aureus isolates
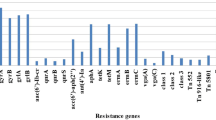
Since every S. aureus carries several virulence genes, but here 19/32 strains carried at least one of the small chosen set of virulence genes (Fig. 2). Enterotoxin genes were detected in majority (53.13%) of the strains, and sej gene was found in 7 (21.88%), followed by seb, sec and see in 18.75% isolates each, sei (9.38%), seg (3.13%). In addition, both tst gene and pvl gene were detected in 6 (18.75%) isolates. The most prevalent combination was determined to be seb + see and seb + tst in 12.5% of all isolates (Table 3). There were 8 isolates which only encode a virulence gene.

Gel electrophoresis of amplified virulence gene products of S. aureus isolates from goats in Chongqing. M: DL-2000 DNA marker; 1: seb (164 bp); 2: sec (451 bp); 3: see (209 bp); 4: seg (704 bp); 5: sei (630 bp); 6: sej (142 bp); 7: tst (326 bp); 8: pvl (255 bp); 9: amplification of seb in Staphylococcus sciuri
MLST analysis
After MLST analysis, 6 new alleles (aroe-552, aroe-553, glpf-500, pta-440,yqil-482 and yqil-496) and 5 new STs (ST 3431,ST 3440,ST 3444,ST 3445 and ST 3461) were identified and added to the S. aureus MLST database (Table 4). The dominant type of aroE was aroE-552, which was present in 92.31% (12/13) of S. aureus isolates, followed by pta-440 and yqiL-496, which accounted for 69.23% (9/13) and 61.54% (8/13) respectively. The ST3444 was the most common ST type which accounted for 61.54% (8/13) of the S. aureus isolates, followed by ST3445 (2 strains), and ST 3431, ST 3440, ST 3461 which just had one strain.
Clonal complexes (CCs) clustering and phylogenetic analysis of S. aureus STs
eBURST generated 103 groups and 483 singletons. The S. aureus isolates tested in this study were clustered into CC522 containing ST3440 and ST3445, and a further CC with no predicted ancestor containing ST3431 and ST3444, while ST3461 was a singleton (Fig. 3). The phylogenetic relationship of 28 STs, based on concatenated sequences of seven MLST alleles, resulted in two main clusters: clusterI(mainly from goats and sheep) and cluster II (from humans). In clusterI, there were three sub-clusters (SCs) (SC1 containing ST130, 522, 1740, 1742, 1758, 2011, 3440, 3445 and 3461; SC2 containing ST133, 425, 1743, 1780, 1781, 2305 and 2328; SC3 containing ST 398, 1595, 3431 and 3444) (Fig. 4).

eBURST-generated CC522 containing the major ST522 comprising 14 STs. The primary founder (ST522) is colored blue and positioned centrally in the cluster, and subgroup founders are colored yellow; the SLVs were connected by red lines, and the DVLs were connected by blue lines; the areas of each of the circles indicate the prevalence of the ST in the input data. The novel STs in this study were marked by red oval circle

Phylogenetic tree of S. aureus STs using the concatenated sequences of the seven MLST genes mainly from human, goats and sheep. ClusterI: mainly from goats and sheep; Cluster II: mainly from human. In clusterI, the STs divided into three sub-clusters (SCs), and the new STs (marked by red filled circles) obtained in this study were clustered in SC1 and SC3. ST398 (marked by black filled diamond), is associated with animals infection [9]; ST522 (marked by black filled triangle) is associated with goats and sheep [2]; ST133 (marked by black filled square) appears to be an ungulate-animal-specific genotype [3]
Discussion
This is the first study of the prevalence of S. aureus from the nose swabs of apparently healthy goats in Chongqing. Our results were comparable to the carriage frequency of S. aureus in healthy sheep in Tunisia [25] approaching to ~ 45% level, which were higher than reported by other investigators in dairy sheep and goats [26,27,28], but lower than that in goats in Norway and Denmark [22, 29].
It is interesting that all the S. aureus isolates in this study were methicillin-susceptible S. aureus (MSSA) since they were sensitive or medium sensitivity to cefoxitin and mecA tested negative. The majority of S. aureus showed relatively low resistance to vancomycin, ceftriaxone, macrodantin, streptomycin, kanamycin, tetracycline, gentamicin, cefotaxime and cefepime, which is similar to the resistance of S. aureus isolated from sheep and goat in Spain [23], but lower than the resistance of S. aureus from goats in Taiwan [26]. The relatively high sensitivity spectrum of S. aureus from Chongqing is likely due to a limited use of antimicrobials for goats [28].
The infections caused by S. aureus is associated with its virulence factors which allow it to adhere to surface, invade or avoid the immune system, and cause harmful toxic effects to the host [30, 31]. More than half of isolates in this study were tested positive for SE genes, which is similar to other reports in S. aureus from dairy goats [29] and their milk products [32], and higher than the result in goats and cows affected with mastitis [33]. Of the SE genes tested, sej was the most predominantly occurring gene which is different from the detection rates in goats from Taiwan or sheep in Tunisia where there were no sej occurrence [25, 26]. Except for sej, the seb, sec, see, tst and pvl carriage rate of 18.75% is similar to the other reports in S. aureus from goats and their milk products [26, 32, 34, 35] but lower than sec and tst found in S. aureus from dairy goats [29], or in MSSA from sheep [25]. In contrast to the other report [29], although sec and tst showed the same detection rates, they were not always co-detected. In the previous study, see was identified only in S. aureus associated with mastitis in goat [26], the carry level of seb and see in this study are inconsistent with other reports [32, 35] where there were no S. aureus strain harboured seb and see. Pvl is associated with S. aureus causing goat subclinical mastitis [36]. Different from the previous report [25], we found 18.75% isolates pvl-positive.
To understand the molecular characteristics of S. aureus isolates from Chongqing goats, we used MLST typing and found 6 new alleles (aroe-552, aroe-553, glpf-500, pta-440,yqil-482 and yqil-496) and 5 new STs (3431,3440,3444,3445 and 3461). The dominant alleles belonged to aroE was aroE-552, which occurred in most of S. aureus strains, followed by pta-440 and yqiL-496. In the newly found STs, ST3444 was the most common, which occurred in approximately 60% of the S. aureus isolates. It is relatively normal for S. aureus from goats to possess novel alleles or STs in contrast to human or bovine strains, which may be simply due to the investigation of a new population, since most reports have focused on human clinical isolates or bovine mastitis [37].
The eBURST analyses assigned S. aureus isolates to CC522 containing ST3440 and ST3445, and a further CC with no predicted ancestor. The results from phylogenetic analyses showed that STs from goats and sheep were separated from that of human, thus formed two main clusters. Three new STs (ST3440, 3445 and 3461) were sub-grouped in the branch of SC1 containing ST522, the primary founder ST of CC522, which was reported previously from cases of goat mastitis or goat’s milk samples [2, 3, 23]. ST3444, the predominate ST in this study, and ST3431 were sub-clustered into the branch SC3 containing ST398, which is associated with animals infection [9]. In SC2, ST133 was found in variety of animals including cows, goats and sheep [2, 3, 22, 23], and was found to be the most common animal-associated MLST type [3]. Our results confirmed the relative close relationship of ST 3440, ST3445 and ST3461 with ST522, and ST3444, ST3431 with ST398.
Conclusion
This is the first study to report the prevalence rate, antimicrobial resistance profile, virulence genes association and MLST characteristics of S. aureus from goats in Chongqing municipality. This study will help in tracking evolution of S. aureus epidemic strains and proving the methods to control S. aureus in goats in China.
Abbreviations
- CC:
-
Clonal complex
- DLVs:
-
Double locus variants
- ETs:
-
Exfoliative toxins
- LA-MRSA:
-
Livestock-associated methicillin-resistant S. aureus
- MLST:
-
Multilocus sequence typing
- MSSA:
-
Methicillin-susceptible S. aureus
- PCR:
-
Polymerase chain reaction
- SEs:
-
Enterotoxins
- SLVs:
-
Single locus variants
- STs:
-
Sequence types
References
Monecke S, Gavier-Widen D, Hotzel H, Peters M, Guenther S, Lazaris A, Loncaric I, Muller E, Reissig A, Ruppelt-Lorz A, Shore AC, Walter B, Coleman DC, Ehricht R. Diversity of Staphylococcus aureus isolates in European wildlife. PLoS One. 2016;11(12):e168433.
Bar-Gal GK, Blum SE, Hadas L, Ehricht R, Monecke S, Leitner G. Host-specificity of Staphylococcus aureus causing intramammary infections in dairy animals assessed by genotyping and virulence genes. Vet Microbiol. 2015;176(1–2):143–54.
Smyth DS, Feil EJ, Meaney WJ, Hartigan PJ, Tollersrud T, Fitzgerald JR, Enright MC, Smyth CJ. Molecular genetic typing reveals further insights into the diversity of animal-associated Staphylococcus aureus. J Med Microbiol. 2009;58(Pt 10):1343–53.
Spini RG, Ferraris V, Glasman MP, Orofino G, Casanovas A, Debaisi G. Methicillin resistant Staphylococcus aureus community acquired meningitis: a case report. Arch Argent Pediatr. 2014;112(6):e266–8.
Song Z, Gu FF, Guo XK, Ni YX, He P, Han LZ. Antimicrobial resistance and molecular characterization of Staphylococcus aureus causing childhood pneumonia in shanghai. Front Microbiol. 2017;8:455.
El-Ashker M, Gwida M, Tomaso H, Monecke S, Ehricht R, El-Gohary F, Hotzel H. Staphylococci in cattle and buffaloes with mastitis in Dakahlia governorate, Egypt. J Dairy Sci. 2015;98(11):7450–9.
Petersen A, Stegger M, Heltberg O, Christensen J, Zeuthen A, Knudsen LK, Urth T, Sorum M, Schouls L, Larsen J, Skov R, Larsen AR. Epidemiology of methicillin-resistant Staphylococcus aureus carrying the novel mecC gene in Denmark corroborates a zoonotic reservoir with transmission to humans. Clin Microbiol Infect. 2013;19(1):E16–22.
Verkade E, Kluytmans J. Livestock-associated Staphylococcus aureus CC398: animal reservoirs and human infections. Infect Genet Evol. 2014;21:523–30.
Loncaric I, Brunthaler R, Spergser J. Suspected goat-to-human transmission of methicillin-resistant Staphylococcus Aureus sequence type 398. J Clin Microbiol. 2013;51(5):1625–6.
De Martino L, Lucido M, Mallardo K, Facello B, Mallardo M, Iovane G, Pagnini U, Tufano MA, Catalanotti P. Methicillin-resistant staphylococci isolated from healthy horses and horse personnel in Italy. J Vet Diagn Investig. 2010;22(1):77–82.
Lozano C, Aspiroz C, Charlez L, Gomez-Sanz E, Toledo M, Zarazaga M, Torres C. Skin lesion by methicillin-resistant Staphylococcus aureus ST398-t1451 in a Spanish pig farmer: possible transmission from animals to humans. Vector Borne Zoonotic Dis. 2011;11(6):605–7.
EFSA. Assessment of the public health significance of meticillin resistant Staphylococcus aureus (MRSA) in animals and foods. EFSA J. 2009;993:2–3.
Brakstad OG, Aasbakk K, Maeland JA. Detection of Staphylococcus aureus by polymerase chain reaction amplification of the nuc gene. J Clin Microbiol. 1992;30(7):1654–60.
CLSI CALS. Performance standards for antimicrobial SusceptibilityTesting; twenty-second informational supplement; CLSI document M100-S22. Wayne, PA, USA: Clinical and Laboratory Standards Institute; 2013.
Murakami K, Minamide W, Wada K, Nakamura E, Teraoka H, Watanabe S. Identification of methicillin-resistant strains of staphylococci by polymerase chain reaction. J Clin Microbiol. 1991;29(10):2240–4.
Mehrotra M, Wang G, Johnson WM. Multiplex PCR for detection of genes for Staphylococcus aureus enterotoxins, exfoliative toxins, toxic shock syndrome toxin 1, and methicillin resistance. J Clin Microbiol. 2000;38(3):1032–5.
McLauchlin J, Narayanan GL, Mithani V, O'Neill G. The detection of enterotoxins and toxic shock syndrome toxin genes in Staphylococcus aureus by polymerase chain reaction. J Food Prot. 2000;63(4):479–88.
Monday SR, Bohach GA. Use of multiplex PCR to detect classical and newly described pyrogenic toxin genes in staphylococcal isolates. J Clin Microbiol. 1999;37(10):3411–4.
Wang Q, Yang LM, Luo FJ, F MS: The resistance and mecA, qacA/B, pvl gene detection of Staphylococcus aureus. In: 2008 Annual Meeting of Zhejiang Provincial Laboratory Medicine. Hangzhou Zhejiang, China: China Academic Journal Electronic Publishing House; 2008: 5.
Noguchi N, Nakaminami H, Nishijima S, Kurokawa I, So H, Sasatsu M. Antimicrobial agent of susceptibilities and antiseptic resistance gene distribution among methicillin-resistant Staphylococcus aureus isolates from patients with impetigo and staphylococcal scalded skin syndrome. J Clin Microbiol. 2006;44(6):2119–25.
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186(5):1518–30.
Eriksson J, Espinosa-Gongora C, Stamphoj I, Larsen AR, Guardabassi L. Carriage frequency, diversity and methicillin resistance of Staphylococcus aureus in Danish small ruminants. Vet Microbiol. 2013;163(1–2):110–5.
Porrero MC, Hasman H, Vela AI, Fernandez-Garayzabal JF, Dominguez L, Aarestrup FM. Clonal diversity of Staphylococcus aureus originating from the small ruminants goats and sheep. Vet Microbiol. 2012;156(1–2):157–61.
Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596–9.
Gharsa H, Ben SK, Lozano C, Gomez-Sanz E, Klibi N, Ben SR, Gomez P, Zarazaga M, Boudabous A, Torres C. Prevalence, antibiotic resistance, virulence traits and genetic lineages of Staphylococcus aureus in healthy sheep in Tunisia. Vet Microbiol. 2012;156(3–4):367–73.
Chu C, Yu C, Lee Y, Su Y. Genetically divergent methicillin-resistant Staphylococcus aureus and sec-dependent mastitis of dairy goats in Taiwan. BMC Vet Res. 2012;8:39.
Vautor E, Abadie G, Guibert JM, Chevalier N, Pepin M. Nasal carriage of Staphylococcus aureus in dairy sheep. Vet Microbiol. 2005;106(3–4):235–9.
Wang YZ, Wang Y, Han X, Yue H, Tang C. Prevalence and resistance analysis of Staphylococcus aureus in clinical healthy goats. Chin Anim Husb Vet Med. 2011;38(3):167–70.
Mork T, Kvitle B, Mathisen T, Jorgensen HJ. Bacteriological and molecular investigations of Staphylococcus aureus in dairy goats. Vet Microbiol. 2010;141(1–2):134.
Holmes A, Ganner M, McGuane S, Pitt TL, Cookson BD, Kearns AM: Staphylococcus aureus isolates carrying Panton-valentine leucocidin genes in England and Wales: frequency, characterization, and association with clinical disease. J Clin Microbiol. 2005;43(5):2384–90.
Bien J, Sokolova O, Bozko P. Characterization of virulence factors of Staphylococcus aureus: novel function of known virulence factors that are implicated in activation of airway epithelial Proinflammatory response. J Pathog. 2011;2011:601905.
Scherrer D, Corti S, Muehlherr JE, Zweifel C, Stephan R. Phenotypic and genotypic characteristics of Staphylococcus aureus isolates from raw bulk-tank milk samples of goats and sheep. Vet Microbiol. 2004;101(2):101–7.
Da SE, Do CL, Da SN. Detection of the enterotoxins a, B, and C genes in Staphylococcus aureus from goat and bovine mastitis in Brazilian dairy herds. Vet Microbiol. 2005;106(1–2):103–7.
Basanisi MG, Nobili G, Bella GL, Russo R, Spano G, Normanno G, Salandra GL. Molecular characterization of Staphylococcus aureus isolated from sheep and goat cheeses in southern Italy. Small Ruminant Res. 2015;135:17–9.
Xing X, Zhang Y, Wu Q, Wang X, Ge W, Wu C. Prevalence and characterization of Staphylococcus aureus isolated from goat milk powder processing plants. Food Control. 2016;59:644–50.
Unal N, Askar S, Macun HC, Sakarya F, Altun B, Yildirim M. Panton-valentine leukocidin and some exotoxins of Staphylococcus Aureus and antimicrobial susceptibility profiles of staphylococci isolated from milks of small ruminants. Trop Anim Health Prod. 2012;44(3):573–9.
Aires-de-Sousa M, Parente CE, Vieira-da-Motta O, Bonna IC, Silva DA, de Lencastre H. Characterization of Staphylococcus aureus isolates from buffalo, bovine, ovine, and caprine milk samples collected in Rio de Janeiro state, Brazil. Appl Environ Microbiol. 2007;73(12):3845–9.
Acknowledgements
We really appreciate Dr. Shijun Hu, Fangjun Cheng and Cailiang Fan for their practical support, Mr. Youdong Gong and Peng Chen for their assistant in sample collection. We also thank Dr. Narayan C. Rath and Yang Zhou for English-language editing of this manuscript.
Funding
This research is funded by the the Social Undertakings and Livelihood Security Technology Innovation Projects of Chongqing (CSTC2015SHMSZX80020), the Project of Youth Fund of Southwest University (No. 20700926) and Fundamental Research Funds for the Central Universities (XDJK2017D078). The funding bodies had no role in study design, data collection or analysis, decision to publish or preparation of the manuscript.
Availability of data and materials
All datasets supporting our findings are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
ZYZ and MSZ designed the study and wrote the manuscript. MSZ, HXL, HYY, XXL, XYS, and ZYW performed the experiments, analyzed the data, and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animal procedures were reviewed and approved by the Ethics Committee of Southwest University. Consent was verbally approved by owners of the goats to take the nasal swabs.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, Z., Zhang, M., Li, H. et al. Prevalence and molecular characterization of Staphylococcus aureus isolated from goats in Chongqing, China. BMC Vet Res 13, 352 (2017). https://doi.org/10.1186/s12917-017-1272-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-017-1272-4