
Abstract
Background
The use of corticosteroids (e.g., dexamethasone) as treatment for tendinopathy has recently been questioned as higher risks for ruptures have been observed clinically. In vitro studies have reported that dexamethasone exposed tendon cells, tenocytes, show reduced cell viability and collagen production. Little is known about the effect of dexamethasone on the characteristics of tenocytes. Furthermore, there are uncertainties about the existence of apoptosis and if the reduction of collagen affects all collagen subtypes.
Methods
We evaluated these aspects by exposing primary tendon cells to dexamethasone (Dex) in concentrations ranging from 1 to 1000 nM. Gene expression of the specific tenocyte markers scleraxis (Scx) and tenomodulin (Tnmd) and markers for other mesenchymal lineages, such as bone (Alpl, Ocn), cartilage (Acan, Sox9) and fat (Cebpα, Pparg) was measured via qPCR. Cell viability and proliferation was calculated using a MTS Assay. Cell death was measured by LDH assay and cleaved caspase-3 using Western Blot. Gene expression of collagen subtypes Col1, Col3 and Col14 was analyzed using qPCR.
Results
Stimulation with Dex decreased cell viability and LDH levels. Dex also induced a significant reduction of Scx gene expression and a marked loss of fibroblast like cell shape. The mRNA for all examined collagen subtypes was found to be down-regulated. Among non-tendinous genes only Pparg was significantly increased, whereas Acan, Alpl and Sox9 were reduced.
Conclusions
These results indicate a Dex induced phenotype drift of the tenocytes by reducing scleraxis expression. Reduction of several collagen subtypes, but not cell death, seems to be a feature of Dex induced tissue degeneration.
Similar content being viewed by others

Background
Chronic tendon pain (tendinopathy) is a troublesome condition that often affects professional and recreational athletes but can also be found among non-active individuals [1–3]. The histopathological features (tendinosis) are mainly defined as structural degeneration characterized by reduced levels of type 1 collagen and haphazardly increased proliferation of smaller type 3 collagen and increased vascularization [4, 5]. Furthermore, cell proliferation and change in cell shape towards rounded and wavy cells, but also occasional cell death has been reported [4, 5]. In addition, it has been shown that in some areas of the tendon features of lipoid degeneration can occur [4]. Furthermore, adherence of fat rich peritendinous tissues onto the tendon proper has been observed [6, 7]. Calcification is another feature that is frequently present potentially being driven by an abnormal pathway of tendon healing [4, 8]. In areas with high levels of compressive loads fibrocartilaginous metaplasia has been observed [4, 9]. The mechanisms behind the tissue changes are not fully understood. However, there is some evidence that extensive load and the imbalance of molecules such as hormones and neuropeptides are involved [9–11].
Treatment methods for tendinopathies are often applied with low scientific evidence [12] and there are controversies about the use of some of them. Especially the application of glucocorticoids, e.g., the commonly used substance dexamethasone (Dex), is still heavily debated and questioned. Glucocorticoids in general are accepted as effective treatments for many inflammatory conditions [13]. For many years tendinopathies were considered as non-inflammatory and degenerative condition (“tendinosis”) by several authors as no signs of a classical inflammation was found in the tendon proper [14, 15]. However, recent studies using advances techniques have suggested that tendon degeneration may be an active process with involvement of aspects of inflammation-mediated responses especially in the early stages [16–18]. Clinical studies on the use of glucocorticoids for tendinopathies have reported short-term pain relief but poor long-term effects including a weakening of tendon structure on the longer perspective [19, 20]. In fact, many reports about spontaneous, often non-traumatic tendon ruptures following the use of these substances have been published [21–28].
In vitro studies have shown that exposure to glucocorticoids has negative effects on tendon cells [29]. It can lead to a reduction in cell viability, proliferation and reduced production of total collagen particularly collagen 1, and proteoglycans [30–35]. These effects are thought to weaken the tendon structure indicated by decreased mechanical properties [29, 36] and may eventually lead to higher risks for tendon ruptures. Conflicting data are available on whether apoptosis is a feature involved in this process [32, 37, 38]. Furthermore it is unclear if all collagen subtypes are affected by dexamethasone in the same way or if it is restricted to the collagen I subtype. As tendinopathy is indicated by an increased collagen 3 production [5] it is important to know how the expression of this subtype responds to glucocorticoids. The effect on collagen type 14, often found in areas of high mechanical stress [39, 40], needs to be evaluated as well.
Recently it has been reported that dexamethasone can influence the differentiation of tendon stem cells in vitro and that it thus can have an impact on the phenotype of cells towards non-tendinous like cells [41–43]. However, no attention has been paid on the potential effect on the expression of tendon specific markers in differentiated primary tendon cells, tenocytes. This is a drawback since this cell population makes up the vast majority of cells in the tendon. The primary purpose of this study was therefore to analyze the expression of tenocyte specific markers scleraxis and tenomodulin in response to Dexamethasone stimulation. Furthermore, the effect on the occurrence of cell death and the expression of collagen subtypes 1, 3 and 14 were evaluated.
Methods
Material and cell culture
Tendon cells used in the present study were derived from plantaris tendon samples from patients (n = 7; 5 men, 2 women; mean age 46 years) with chronic co-existing midportion Achilles and plantaris tendinopathy [44]. All patients had pain duration of at least 3 months and verified tendinopathies as shown by clinical examination including Ultrasound and Colour Doppler (US/CD). Exclusion criteria were acute and chronic inflammatory diseases, previous intratendinous treatments and partial tears in the Achilles tendon.
The tissue was obtained in the course of a previously described surgical procedure that includes plantaris tendon removal and Achilles tendon scraping [7, 45]. The isolation and culturing of primary tendon cells was carried out according to a protocol that has been routinely used in our laboratory [46]. Cells were cultured in DMEM supplemented with 10 % fetal bovine serum (FBS; Invitrogen, code: 16000), 1 % pen-strep (Invitrogen; code: 15140) and 0.2 % L-glutamine (Invitrogen; code: 25030) at 37 °C in a humidified atmosphere of 5 % CO2.
The cells in this study were used in passages 2–4. Prior to stimulation with dexamethasone cells were serum-starved for 24 h with 1 % FBS in DMEM. All experiments (see below) were performed at least three times and cells from at least two different patients were used.
Ethical considerations
The principles expressed in the Declaration of Helsinki were followed. A written informed consent was received from all patients included in this study. Ethical approvals were obtained from the Ethical Committee at the Medical Faculty of Umeå University, and the Regional Ethical Review Board in Umeå (dnr 04-157 M; 2011-83-32 M).
Immunocytochemistry
In order to characterize the primary tendon cells in vitro immunocytochemistry (ICC) was performed. Antibodies were directed towards hematopoietic progenitor cell antigen CD34 (1:25; Santa Cruz; code: sc9095), collagen 1 (COL1) (1:50; Abcam; code: ab34710), collagen 3 (COL3) (1:50; Abcam; code: ab7778), octamer-binding transcription factor 4 (OCT4) (1:50; Abcam; code: ab27985), scleraxis (SCX) 1:50; Abcam; code: ab58655), tenomodulin (TMD) 1:50; Abcam; code: ab203676).
For the procedure 15,000 cells/well were seeded on 8-well chamber slides overnight. The following day cells were initially fixed in 3.7 % paraformaldehyde in 1× PBS for 15 min. Then they were washed 4 times for 1 min in 1× PBS. After that cells were blocked with normal serum (1:20 in 1× PBS) for 15 min and incubated with the primary antibody for 60 min at 37 °C. Subsequently they were washed and blocked again, and incubated with the secondary antibody for 30 min at 37 °C. Eventually another washing step was performed before the slides were mounted using ProLong® Diamond Antifade Mountant with DAPI (Life Technologies; code: P36962). In order to evaluate the specificity of the immunofluorescence a control staining, where the primary antibody was replaced with 1× PBS, was performed. The immunoreactions were analyzed via a Zeiss Axioskop 2 plus microscope equipped with epifluorescence and an Olympus DP70 digital camera. The figure montages were created with Photoshop CS5 Extended software.
When using primary antibodies from rabbits, swine normal serum (1:20; Jackson I.R; code: 014-000-121) was used for blocking and swine anti-rabbit secondary antibody conjugated with tetramethylrhodamine isocyanate (TRITC) (1:40; DAKO; code: R0156) was applied. For goat antibodies, donkey normal serum (1:20; Jackson I.R; code: 017-000-121) and donkey anti goat secondary antibody conjugated with fluorescein isothiocyanate (FITC) (1:50; Jackson I.R.; code: 705-095-147) were used.
Stimulation with dexamethasone (Dex)
Dexamethasone (Sigma; code: D4902) was solved in pure ethanol and diluted into concentrations of 1 mM, 10 mM, 100 mM and 1000 mM. For exposure the Dex solutions were applied 1:1000 in 1 % FBS in DMEM (2 μl Dex + 2 ml 1 % FBS in DMEM). Thus the final concentrations of Dex were 1 nM, 10 nM, 100 nM and 1000 nM. The durations of stimulation (time points) were 6 h, 12 h, 24 h, 48 h, 72 h, 96 h and 120 h. The control, unstimulated cells, were exposed to pure ethanol using the same volume (2 μl ethanol + 2 ml 1 % FBS in DMEM).
Measuring cell death (LDH assay)
Cell death in response to the Dex exposure was measured by a lactate dehydrogenase (LDH) assay from Promega (code: G1780). At each time point supernatant was collected and then immediately stored at -20 °C. For the analysis 50 μl of supernatant was pipetted into a 96-well plate, then mixed with 50 μl reconstituted substrate mix and incubated for 30 min in a light protected environment under shaking. After that 50 μl of stop solution was applied and the absorbance was read at 490 nm using a plate reader. LDH was measured at all time points and all concentrations described above.
Measuring cell viability (MTS assay)
The effect of dexamethasone on the proliferation and viability of tendon cells was measured using a MTS assay (CellTiter 96® Aqueous One Solution Cell Proliferation Assay; code: G3581). For this purpose tendon cells were seeded in a 96 well plate overnight at a density of 5,000/well. For analysis MTS reagent (20 μl per 100 μl media) was added and incubated for 4 h at 37 °C, 5 % CO2. The amount of formazan produced by cellular reduction of MTS, was measured with a micro-plate reader and the absorbance of 490 nm. MTS was measured at all time points and all concentrations described above.
RNA isolation, reverse transcription, and qPCR
For analyzing gene expression qPCR was performed. Genes related to collagens (Col1, Col3, Col14) were analyzed. Furthermore markers that are tenocyte specific (Scx, Tnmd) and those characterizing non-tendinous cells (bone: Alpl, Ocn; cartilage: Acan, Sox9; fat: Pparg, Cebpα) were looked at. All applied probes and primers are listed in Tables 1 and 2.
Initially, RNA was isolated using an extraction kit (Qiagen; code: 74106). The extracted RNA was then reverse transcribed into cDNA via a High Capacity cDNA Reverse Transcription kit (Applied Biosystems; code: 4368813). The settings for the conversion which was performed on a thermal cycler (Eppendorf Mastercycler EP Gradient S) were as follow: 10 min at 25 °C, 120 min at 37 °C, 5 min at 85 °C.
For the quantitative PCR (qPCR) either TaqMan fast universal PCR mastermix (Applied Biosystems; code: 4352042) or SYBR Green fast universal PCR mastermix (Applied Biosystems; code: 4385612) was used. For each of the amplifications, 40 ng of cDNA was taken and analyzed in technical dublicates.
The amplification was performed in a ViiA7 Real-Time PCR system (Applied Biosystems). The expression levels of genes were calculated in relation to the endogenous control β-actin (Actb) for the Taqman probes and Gapdh when using SYBR Green. For more practical details see [46, 47]. mRNA was analyzed after stimulation with 100 nM Dex after 12 h, 24 h, 48 h and 72 h. For Scx and Tnmd all concentrations (1nM, 10nM, 100nM, 1000nM) were used.
Western blot
Cells were washed in sterile 1× PBS and then stored in -80 °C until all time point were collected. On the day of analyses cells were scraped in lysis buffer (RIPA) supplemented with protease inhibitor (1:200), put in an Eppendorf tube, vortexed and then incubated on ice for 30–60 min. After that the tubes were centrifuged to remove the cell debris. The supernatant was taken and analyzed for concentrations of total proteins using Protein Assay Dye Reagent Concentrate (Bio-Rad; code: 500–0006). Bovine Albumin Serum (BSA; Sigma; code: A9647) was used as a standard. Before loading onto a SDS-PAGE gel, in similar protein concentrations, the samples were boiled in 2× Lammeli buffer (Bio-Rad; code: 161–0737) supplemented with beta-mercaptoethanol. After the run (150 V) the proteins were transferred to a polyvinylidene fluoride transfer membrane for 1 h at 100 V. The membrane was then blocked with either 5 % BSA or 5 % non-fat milk powder in 1× TBS-T for 60 min and eventually incubated with the primary antibody at 4 °C overnight. Next day the membrane was washed in TBS-T (3×5 min) and after that incubated with the secondary antibody at room temperature for 60 min. After the final wash the membranes were treated with chemiluminescent HRP substrate (GE Healthcare; code: RPN2232) for 5 min and then visualized using Odyssey® Fc imaging system (LI-COR, Lincoln, NE, USA).
Antibodies were directed towards cleaved caspase 3 (cCasp3) (Cell Signaling; code: 9664), COL 1 (Cell Signaling; code: 9496), SCX (Abcam; code: ab58655) and TMND (Abcam; code: ab203676). Stimulation with Dex was performed in a concentration of 100 nM for 12 h, 24 h, 48 h and 72 h.
Oil Red O staining
For the detection of adipocytes Oil Red O staining was performed. Before that cells were stimulated with Dex every second day for 21 days and media containing Dex (100 nM) was replaced every second day.
Cells were initially fixed in 3.7 % paraformaldehyde for 30–40 min. After that they were washed with 1× PBS (3x5 min) and rinsed in water twice. After that Oil Red O Solution was applied for 50 min. Cells were then washed with water and eventually stained with hematoxylin (10 min).
Statistical analysis
Data were statistically analyzed by using PASW Statistics 18 (SPSS). One-way analysis of variance (ANOVA), followed by the Bonferroni post-hoc test, and independent t-tests were used. All results were successfully reproduced at least twice and performed in triplicates and technical duplicates. Significance levels were predetermined at p < 0.05.
Results
Primary tendon cells express tenocyte markers
The primary cells revealed immunoreactions for COL 1, SCX, TNMD and to some extent COL 3 (Fig. 1a-d). No, or very little, reactions were seen for stemness marker OCT4 and endothelial marker CD34 (Fig. 1e, f).

Primary tendon cells stained for COL 1 (a), COL 3 (b), TNMD (c), SCX (d), OCT4 (e) and CD34 (f). Immunoreactions are seen for COL1, COL3 (arrows), TNMD and SCX (arrows). No or very little reactions can be seen for OCT4 and CD34
Decrease of cell death, cell viability and proliferation
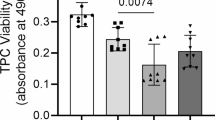
The exposure of Dex leads to a dose-depended reduction of cell viability after 72 h (Fig. 2a). Similar trends could be observed also at earlier time point (not shown).

Effects of Dex on cell viability (MTS) and cell death (LDH) levels. A significant reduction in cell viability (a) and LDH (b) after 72 h of Dex treatment is found. Microscopic pictures of stimulated cells (7 days) stained with Oil Red solution and hematoxylin show lower cell numbers and a loss of fibroblastic appearance after treatment with Dex (c). *** p < 0.001
The LDH assay showed a dose-depended reduction of cell death after 72 h (Fig. 2b) with similar trends for earlier time points. There was no presence of cleaved caspase-3 (not shown) after 24 h. The reduced number of cells is also seen macroscopically (Fig. 2c).
Decreased Scx gene expression after Dex treatment
The expression of scleraxis (Scx) mRNA was reduced (Fig. 3a, b) after Dex treatment at all time points examined, most prominently after 24 h. Gene expression for Tnmd was very low and not considered for analysis.

Results of Dex (100 nM) exposure on the relative Scx gene expression and the expression of non-tendinous genes. A significant down regulation of Scx mRNA is seen (a) and this down regulation is consistent over a 48 h time course (b). Acan, Sox9 and Alpl expression is reduced (c) and Pparg is up-regulated in cells exposed to Dex. *p < 0.05; ** p < 0.01; *** p < 0.001; n.s. (non-significant)
Effects of Dex on tendon cell (tenocyte) phenotype
Among the analyzed non-tenocyte genes, only the fat marker Pparg was significantly up-regulated on mRNA level after Dex treatment (Fig. 3c). However, Oil red O staining after 21 days of stimulation did not show any presence of adipocytes in the culture (not shown).
Acan, Sox9 and Alpl were found to be decreased (Fig. 3). The gene expression of Ocn and Cebpα was very low in both groups.
Stimulated cells exhibited a different shape than untreated cells. They lost their typical elongated fibroblastic appearance (Fig. 2c).
Decreased expression of collagens
Dex stimulation resulted in decreased expression of Col1, Col3 and Col14 on mRNA level after 24 h (Fig. 4a), and similar results for other examined time points (24 h, 48 h, 72 h, 96 h). COL 1 was found to be reduced on protein level as well (Fig. 4b).

Results of Dex (100 nM) exposure on the expression of collagens. mRNA of collagen subtypes Col 1, Col 3 and Col 14 is significantly down-regulated after 24 h of Dex exposure (a) . COL 1 protein is decreased after 24 h and 48 h of Dex exposure (b). * p < 0.05; *** p < 0.001
Discussion
This is to our knowledge the first study evaluating tenocyte marker expression in response to exposure of the corticosteroid Dexamethasone (Dex) in primary tendon cells. Our results suggest that Dex leads to a marked reduction of scleraxis, a transcription factor that has been reported to be essential for tendon development and differentiation [48]. At the same time we found that the tendon cells lost their typical fibroblastic shape during the stimulation. This indicates a potential phenotype change of the tenocytes as a result of Dex. When evaluating the expression of non-tendinous markers, we found that the adipocyte marker PPARG was up-regulated. However, staining for adipocytes did not show a presence of adipocytes within 21 days of stimulation. That is not very surprising since studies have shown that compared to tendon stem cells, primary cells have a very low potential to differentiated into cartilage-, fat- and bone related cells [49]. Studies on tendon stem cells have reported a phenotype change towards fat cells via reduction of scleraxis [41–43]. However, clone formation experiments prior to the present study have indicated that the cells in culture in the present study are mature tendon cells and that the amount of possible stem cells was very little. Thus, at least the marked reduction of scleraxis must be related to changes occurring in primary matured tendon cells. In fact, not all processes of tenocytes differentiation are fully understood yet. Therefore, the reduction of scleraxis accompanied with the change in cell shape in response to Dex could be a sign of reduced tenocyte phenotype, which may potentially affect the tendon. More studies are needed on how this scleraxis reduction affects the cell on a downstream direction.
Another observation of this study is a decrease in cell viability. This has been reported by other studies before [30–32]. Surprisingly, this reduction was not associated with an increase of cell death, as indicated by the reduction of LDH levels in our study. Furthermore, we did not find cleaved caspase-3 by Western Blot. This may indicate that apoptosis/necrosis is not a feature of Dex exposure as previously reported by Wong et al. on primary patellar tendon cells [32]. In fact, there are conflicting data in the literature [32, 37, 38]. These discrepancies might be explained by different concentrations of Dex and the use of cells from different tendons in these studies. In our study, we did not find any signs of increased cell death processes in cells derived from plantaris tendinopathy tendons, with different concentrations (1 nM-1000 nM) of Dex treatment within 5 days. However, it should also be mentioned that LDH does not necessarily detect apoptosis. Necrotic processes are even more likely to be detected since it measures plasma permeability. Based on our results, it rather seems that the turnover in general, i.e., both proliferation and cell death (necrosis/apoptosis), is lower in stimulated cells as both cell viability and cell death is decreased. That doesn’t mean that there is a general decrease in cell death as these results are not normalized. In fact, Poulsen and co-workers have suggested that glucocorticoids can cause senescence in tendon cells, which can explain the reduced cell viability [50].
It is also known that patients with Cushing syndrome (CS) resulting from chronic exposure to excessive circulating levels of glucocorticoids have major alterations in the tendon structure and/or tendon function [51]. Despite based on only a few studies, it shows that chronic use of glucocorticoids harms the tendon even if used systemically.
The third finding of the present work was that Dex not only reduced the expression of collagen 1 but also mRNA of collagen 3 and 14. Previous studies have shown that collagen 1, the main collagen in tendons, is reduced after exposure to corticosteroids [30]. Collagen 3, a smaller collagen, is another collagen type in tendons and known to be up-regulated in the process of tendinopathy [5]. Also collagen 14 is present in tendons [39]. Type 14 collagen is often found in areas of high mechanical stress [40]. The reduction of all examined collagen subtypes indicates that Dex may lead to a weakening of tendon structure. It is therefore not surprising that spontaneous ruptures have been linked to injections of corticosteroids in tendons [21–28]. Our results give a plausible molecular explanation for that.
A drawback of this study is that it is unclear if the Dex concentrations used in vitro are representative for the in vivo situation in the tendon when it is clinically used. However, we applied a wide range of concentrations and stack to those used in previous studies. Another issue is that most mRNA and protein analyses were solely performed after 100nM Dex exposure. The effect of other concentrations was only measured for MTS and LDH. We had tested other concentrations prior to these experiments. Based on the results of these and the fact that 100nM Dex had the highest impact on cell viability we decided to use it for further experiments.
Conclusion
Overall, the results of our study suggest that Dex reduces the tenocyte phenotype via reduction of Scx expression, decreased cell turnover and decreased expression of collagen 1, 3, 14 in primary tendon cells. This study suggests that Dex may weaken the tendon structure in vivo.
Abbreviations
- Acan:
-
Aggrecan
- Actb:
-
β-actin
- Alpl:
-
Alkaline phosphatase
- Cebpa:
-
CCAAT/enhancer-binding protein alpha
- Col1:
-
Collagen type 1
- Col14:
-
Collagen type 14
- Col3:
-
Collagen type 3
- CS:
-
Christoph Spang
- Dex:
-
Dexamethasone
- Gapdh:
-
Glyceraldehyde 3-phosphate dehydrogenase
- JC:
-
Jialin Chen
- LDH:
-
Lactate dehydrogenase
- LJB:
-
Ludvig J Backman
- Ocn:
-
Osteocalcin
- Pparg:
-
Peroxisome proliferator-activated receptor γ
- Scx:
-
Scleraxis
- Sox9:
-
SRY-box 9
- Tnmd:
-
Tenomodulin
References
Rolf C, Movin T. Etiology, histopathology, and outcome of surgery in achillodynia. Foot Ankle Int. 1997;18:565–9.
de Jonge S, van den Berg C, de Vos RJ, et al. Incidence of midportion Achilles tendinopathy in the general population. Br J Sports Med. 2011;45:1026–8.
Albers IS, Zwerver J, Diercks RL, et al. Incidence and prevalence of lower extremity tendinopathy in a Dutch general practice population: a cross sectional study. BMC Musculoskelet Disord. 2016;17:16.
Khan KM, Cook JL, Bonar F, et al. Histopathology of common tendinopathies. Update and implications for clinical management. Sports Med. 1999;27:393–408.
Scott A, Backman L, Speed C. Tendinopathy-Update on Pathophysiology. J Orthop Sports Phys Ther. 2015;45:833–41.
Spang C, Harandi VM, Alfredson H, Forsgren S. Marked innervation but also signs of nerve degeneration in between the Achilles and plantaris tendons and presence of innervation within the plantaris tendon in midportion Achilles tendinopathy. J Musculoskelet Neuronal Interact. 2015;15:197–206.
Masci L, Spang C, van Schie HT, Alfredson H. How to diagnose plantaris tendon involvement in midportion Achilles tendinopathy - clinical and imaging findings. BMC Musculoskelet Disord. 2016;17:97.
Oliva F, Via AG, Maffulli N. Physiopathology of intratendinous calcific deposition. BMC Med. 2012;10:95.
Docking S, Samiric T, Scase E, et al. Relationship between compressive loading and ECM changes in tendons. Muscles Ligaments Tendons J. 2013;3:7–11.
Oliva F, Piccirilli E, Berardi AC, et al. Hormones and tendinopathies: the current evidence. Br Med Bull. 2016;117:39–58.
Danielson P. Reviving the “biochemical” hypothesis for tendinopathy: new findings suggest the involvement of locally produced signal substances. Br J Sports Med. 2009;43:265–8.
Skjong CC, Meininger AK, Ho SS. Tendinopathy treatment: where is the evidence? Clin Sports Med. 2012;31:329–50.
Buttgereit F, Spies CM, Bijlsma JW. Novel glucocorticoids: where are we now and where do we want to go? Clin Exp Rheumatol. 2015;33:S29–33.
Maffulli N, Khan KM, Puddu G. Overuse tendon conditions: time to change a confusing terminology. Arthroscopy. 1998;14:840–3.
Khan KM, Cook JL, Kannus P, Maffulli N, Bonar SF. Time to abandon the “tendinitis” myth. BMJ. 2002;324(7338):626–7.
Al-Sadi O, Schulze-Tanzil G, Kohl B, et al. Tenocytes, pro-inflammatory cytokines and leukocytes: a relationship? Muscles Ligaments Tendons J. 2012;1:68–76.
Rees JD, Stride M, Scott A. Tendons-time to revisit inflammation. Br J Sports Med. 2014;48:1553–7.
Del Buono A, Oliva F, Osti L, et al. Metalloproteases and tendinopathy. Muscles Ligaments Tendons J. 2013;3:51–7.
Coombes BK, Bisset L, Vicenzino B. Efficacy and safety of corticosteroid injections and other injections for management of tendinopathy: a systematic review of randomised controlled trials. Lancet. 2010;376:1751–67.
Scott A, Khan KM. Corticosteroids: short-term gain for long-term pain? Lancet. 2010;376:1714–5.
Haines JF. Bilateral rupture of the Achilles tendon in patients on steroid therapy. Ann Rheum Dis. 1983;42:652–4.
Baruah DR. Bilateral spontaneous rupture of the Achilles tendons in a patient on long-term systemic steroid therapy. Br J Sports Med. 1984;18:128–9.
Newnham DM, Douglas JG, Legge JS, Friend JA. Achilles tendon rupture: an underrated complication of corticosteroid treatment. Thorax. 1991;46:853–4.
Kotnis RA, Halstead JC, Hormbrey PJ. Atraumatic bilateral Achilles tendon rupture: an association of systemic steroid treatment. J Accid Emerg Med. 1999;16:378–9.
Madsen BL, Noer HH. Simultaneous rupture of both peroneal tendons after corticosteroid injection: operative treatment. Injury. 1999;30:299–300.
Smith AG, Kosygan K, Williams H, Newman RJ. Common extensor tendon rupture following corticosteroid injection for lateral tendinosis of the elbow. Br J Sports Med. 1999;33:423–4.
Mills SP, Charalambous CP, Hayton MJ. Bilateral rupture of the extensor pollicis longus tendon in a professional goalkeeper following steroid injections for extensor tenosynovitis. Hand Surg. 2009;14:135–7.
Zhang B, Hu ST, Zhang YZ. Spontaneous rupture of multiple extensor tendons following repeated steroid injections: a case report. Orthop Surg. 2012;4:118–21.
Dean BJ, Lostis E, Oakley T, et al. The risks and benefits of glucocorticoid treatment for tendinopathy: a systematic review of the effects of local glucocorticoid on tendon. Semin Arthritis Rheum. 2014;43:570–6.
Kempka G, Ahr HJ, Ruther W, Schluter G. Effects of fluoroquinolones and glucocorticoids on cultivated tendon cells in vitro. Toxicol In Vitro. 1996;10:743–54.
Tsai WC, Tang FT, Wong MK, et al. Decreased expression of proliferating cell nuclear antigen is associated with dexamethasone inhibition of the proliferation of rat tendon fibroblasts. J Rheumatol. 2002;29:2397–402.
Wong MW, Tang YY, Lee SK, et al. Effect of dexamethasone on cultured human tenocytes and its reversibility by platelet-derived growth factor. J Bone Joint Surg Am. 2003;85-A:1914–20.
Wong MW, Tang YY, Lee SK, Fu BS. Glucocorticoids suppress proteoglycan production by human tenocytes. Acta Orthop. 2005;76:927–31.
Wong MW, Lui WT, Fu SC, Lee KM. The effect of glucocorticoids on tendon cell viability in human tendon explants. Acta Orthop. 2009;80:363–7.
Carofino B, Chowaniec DM, McCarthy MB, et al. Corticosteroids and local anesthetics decrease positive effects of platelet-rich plasma: an in vitro study on human tendon cells. Arthroscopy. 2012;28:711–9.
Olchowik G, Siek E, Tomaszewska M, Tomaszewski M. The evaluation of mechanical properties of animal tendons after corticosteroid therapy. Folia Histochem Cytobiol. 2008;46:373–7.
Hossain MA, Park J, Choi SH, Kim G. Dexamethasone induces apoptosis in proliferative canine tendon cells and chondrocytes. Vet Comp Orthop Traumatol. 2008;21:337–42.
Sendzik J, Shakibaei M, Schafer-Korting M, et al. Synergistic effects of dexamethasone and quinolones on human-derived tendon cells. Int J Antimicrob Agents. 2010;35:366–74.
Young BB, Gordon MK, Birk DE. Expression of type XIV collagen in developing chicken tendons: association with assembly and growth of collagen fibrils. Dev Dyn. 2000;217:430–9.
Ansorge HL, Meng X, Zhang G, et al. Type XIV Collagen Regulates Fibrillogenesis: Premature collagen fibril growth and tissue dysfunction in null mice. J Biol Chem. 2009;284:8427–38.
Zhang J, Keenan C, Wang JH. The effects of dexamethasone on human patellar tendon stem cells: implications for dexamethasone treatment of tendon injury. J Orthop Res. 2013;31:105–10.
Chen W, Tang H, Zhou M, et al. Dexamethasone inhibits the differentiation of rat tendon stem cells into tenocytes by targeting the scleraxis gene. J Steroid Biochem Mol Biol. 2015;152:16–24.
Chen W, Tang H, Liu X, et al. Dickkopf1 Up-Regulation Induced by a High Concentration of Dexamethasone Promotes Rat Tendon Stem Cells to Differentiate Into Adipocytes. Cell Physiol Biochem. 2015;37:1738–49.
Spang C, Alfredson H. Ferguson et al. The plantaris tendon in association with mid-portion Achilles tendinosis: tendinosis-like morphological features and presence of a non-neuronal cholinergic system. Histol Histopathol. 2013;28:623–32.
Masci L, Spang C, van Schie HTM, et al. Achilles tendinopathy-do plantaris tendon removal and Achilles tendon scraping improve tendon structure? A prospective study using ultrasound tissue characterization. BMJ Open Sport Exerc Med. 2015;1:e000005.
Backman LJ, Fong G, Andersson G, et al. Substance P is a mechanoresponsive, autocrine regulator of human tenocyte proliferation. PLoS One. 2011;6:e27209.
Chen J, Zhang W, Liu Z, et al. Characterization and comparison of post-natal rat Achilles tendon-derived stem cells at different development stages. Sci Rep. 2016;6:22946.
Liu H, Zhu S, Zhang C, et al. Crucial transcription factors in tendon development and differentiation: their potential for tendon regeneration. Cell Tissue Res. 2014;356:287–98.
Zhang J, Wang JH. Characterization of differential properties of rabbit tendon stem cells and tenocytes. BMC Musculoskelet Disord. 2010;11:10.
Poulsen RC, Watts AC, Murphy RJ, et al. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: in vivo and in vitro evidence. Ann Rheum Dis. 2012;73:1405–13.
Galdiero M, Auriemma RS, Pivonello R, et al. Cushing, acromegaly, GH deficiency and tendons. Muscles Ligaments Tendons J. 2014;4:329–32.
Acknowledgment
The authors would like to thank Dr. Paul Kingham and Dr. Peyman Kelk for their valuable input and help with issues related to cell characterization and differentiation. Furthermore we want to thank Juha Pillti for his input related to the qPCR experiments. We also thank Prof. Håkan Alfredson for collection of tendon biopsies and Lotta Alfredson for co-ordinating.
Funding
This work was financed by Umeå University.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request
Authors’ contributions
All authors were involved in the design of the study and in the analysis and discussion of the results. CS performed the experiments, created the figures and graphs, and wrote the first draft of the manuscript. JC did parts of the experiment and gave practical advice, and participated in the manuscript writing. LJB did parts of the experiments and participated in the manuscript writing. All authors read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The principles expressed in the Declaration of Helsinki were followed. A written informed consent was received from all patients included in this study. Ethical approvals were obtained from the Ethical Committee at the Medical Faculty of Umeå University, and the Regional Ethical Review Board in Umeå (dnr 04-157 M; 2011-83-32 M).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Spang, C., Chen, J. & Backman, L.J. The tenocyte phenotype of human primary tendon cells in vitro is reduced by glucocorticoids. BMC Musculoskelet Disord 17, 467 (2016). https://doi.org/10.1186/s12891-016-1328-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12891-016-1328-9