
Abstract
Background
There is emerging evidence that there are shared genetic, environmental and developmental risk factors in psychiatry, that cut across traditional diagnostic boundaries. With this background, the Discovery biology of neuropsychiatric syndromes (DBNS) proposes to recruit patients from five different syndromes (schizophrenia, bipolar disorder, obsessive compulsive disorder, Alzheimer’s dementia and substance use disorders), identify those with multiple affected relatives, and invite these families to participate in this study. The families will be assessed: 1) To compare neuro-endophenotype measures between patients, first degree relatives (FDR) and healthy controls., 2) To identify cellular phenotypes which differentiate the groups., 3) To examine the longitudinal course of neuro-endophenotype measures., 4) To identify measures which correlate with outcome, and 5) To create a unified digital database and biorepository.
Methods
The identification of the index participants will occur at well-established specialty clinics. The selected individuals will have a strong family history (with at least another affected FDR) of mental illness. We will also recruit healthy controls without family history of such illness. All recruited individuals (N = 4500) will undergo brief clinical assessments and a blood sample will be drawn for isolation of DNA and peripheral blood mononuclear cells (PBMCs). From among this set, a subset of 1500 individuals (300 families and 300 controls) will be assessed on several additional assessments [detailed clinical assessments, endophenotype measures (neuroimaging- structural and functional, neuropsychology, psychophysics-electroencephalography, functional near infrared spectroscopy, eye movement tracking)], with the intention of conducting repeated measurements every alternate year. PBMCs from this set will be used to generate lymphoblastoid cell lines, and a subset of these would be converted to induced pluripotent stem cell lines and also undergo whole exome sequencing.
Discussion
We hope to identify unique and overlapping brain endophenotypes for major psychiatric syndromes. In a proportion of subjects, we expect these neuro-endophenotypes to progress over time and to predict treatment outcome. Similarly, cellular assays could differentiate cell lines derived from such groups. The repository of biomaterials as well as digital datasets of clinical parameters, will serve as a valuable resource for the broader scientific community who wish to address research questions in the area.
Similar content being viewed by others


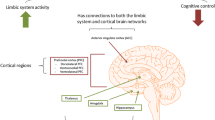
Background
Severe mental illnesses are a major source of morbidity and disability, with about 2–3% of the population at risk for developing these disorders [1]. These illnesses usually begin in early adult life, with almost 75% of patients developing symptoms by age 24 [2], and often have chronic course. Our current understanding of these diseases suggests that the risk factors that underlie these illnesses have antecedents in early life [3, 4]. In contrast to other chronic medical illnesses, such as cardiovascular diseases or cancers, people with mental disorders become ill at the prime of life and intellectual growth, thus impacting lifespan, quality of life and career [5]. These disorders lead to significant mortality and, morbidity, which requires support at several levels (pharmacological and psycho-social interventions, and rehabilitation). Most interventions are targeted towards amelioration of particular symptoms, as the primary disease processes remain ill understood; and unmet clinical needs remain high [6, 7].
Traditional clinical classification systems conceptualize the psychiatric disorders as a group of discrete syndromes that are independent of each other. For example, schizophrenia, bipolar disorders and other psychotic disorders are considered to differ from each other, based on their unique clinical characteristics, course, family history and treatment response [8]. However, there is now accumulating evidence that there exist overlapping genetic, environmental and developmental factors, cutting across these diagnostic boundaries [9,10,11,12,13,14,15]. Cross-disorder consortia studies have attempted to understand the genetic basis of these overlaps and the shared pathophysiology and have found high rates of heritability and co-heritability [16,17,18]. Many of the genes (and pathways) identified suggest that differences in neural development and connectivity in early life are critical to their pathogenesis. These variations, coupled with epigenetic dysregulation in the brain; influenced by various environmental factors, acting at various points in time during critical neurodevelopmental time windows, influence the onset, and progression, of illness [19]. Disease associated biological signatures are often evident in at-risk individuals of many severe adolescence onset psychiatric disorders (e.g. bipolar disorder (BD) [20, 21], schizophrenia [22], substance use disorders (SUD) [23,24,25], obsessive compulsive disorder (OCD) [26]) before being manifest as a clinically recognizable syndrome. There is emerging evidence that this also holds true for late onset diseases affecting the brain, and differences in hippocampal structure are evident in childhood or adolescence in persons at risk for Alzheimer’s dementia (AD) [27].
Given these observations, it is necessary that attempts to understand the biology of these disorders considers their genetic basis, differences in neurodevelopment, as well as the overlapping nature of the individual psychiatric syndromes. One way to account for these factors would be to prospectively follow-up affected and unaffected individuals, whose family history suggests an elevated risk of developing disease (based on both clinical information and genetic analysis) from a stage when they are asymptomatic, so that clinical investigations can capture the evolution of alterations in brain function. Such a strategy combined with modern human genetics and analysis of cellular function in brain cells using ‘disease in dish’ models should help us understand the cellular and molecular underpinnings psychiatric illness.
The Discovery biology of neuropsychiatric syndromes [DBNS] is one such research initiative in Bangalore, India, built on a collaboration between clinicians and scientists at the National Institute of Mental Health and Neuro Sciences (NIMHANS), the National Center for Biological Sciences (NCBS), and the Institute for Stem Cell Biology and Regenerative Medicine (InStem). It aims to understand the developmental trajectories and basic biology of these major psychiatric disorders. The study will use multiple techniques (brain imaging, psycho-physics, neuropsychology, next generation sequencing, cellular models), and in-depth clinical assessments of participants in a cohort of multiple affected families with a strong family history of mental illness. The facilities created, and the data sets, will provide a resource for future research to help answer questions with implications for basic neuroscience as well as drive translational research with positive impact for clinical psychiatry.
Methods
Rationale
The basic premise of this research initiative is that there exist shared genetic, environmental and developmental factors across the major psychiatric disorders. We aim to identify such overlaps by examining clinical, brain imaging, neurophysiological and neuropsychological measures, as well as clinical histories, across disorders. In addition, we would examine the longitudinal trajectory of these neurobiological measures. We will recruit multiple affected families with patients having one or more of 5 syndromes (schizophrenia, BD, OCD, AD and SUD) so that they broadly represent the major and common psychopathologies. The identification of the index participants will occur at well-established specialty clinics at NIMHANS. Importantly, the index patients chosen will have a strong family history of mental illness. The families will then become part of a single sample, who will be subjected to a uniform set of clinical analyses; and a bio-repository will be set up using cellular material from these individuals. Several endophenotype assessments (neuro-imaging, psycho-physiology, neuropsychology), will be also be performed, as these empirical measures will provide robust and consistent measures of brain function. Familial stress, and psycho-social factors will also be evaluated using structured assessments. It is expected that such deep interrogation of brain networks and sub-cellular networks using molecular and cellular analysis; as well as social and psychological measurements, would lead to better mechanistic understanding of these disorders.
Objectives
Specific objectives of the program are as follows:
Short-term objectives
-
a.
To identify 300 families in whom multiple members (more than 2 affected first degree relatives in a nuclear family) are diagnosed to have a major psychiatric disorder (schizophrenia, BD, OCD, AD and/ or SUD) with structured assessments.
-
b.
To study brain structural [grey (magnetic resonance imaging-MRI) and white matter (diffusion tensor MRI)] abnormalities, resting and task-related functional MRI activity, neuropsychological performance, brain electrical activity and eye movement abnormalities in probands with major psychiatric disorders and their unaffected first-degree relatives (FDR); in comparison with matched healthy controls
-
c.
To identify cellular phenotypes that differentiate between the groups using experiments on cell lines [lymphoblastoid cell lines (LCL), induced pluripotent stem cell (IPS) lines, IPS derived neurons/glia]
-
d.
Create a unified digital database of all the above information.
Long-term objectives (5 years and beyond)
-
e.
To examine the time course and progression of structural/functional brain abnormalities, neuropsychological performance, brain electrical functions and eye movement abnormalities in probands and unaffected FDRs and study their relationship to course of illness and disease conversion.
-
f.
To examine cell lines from good and poor outcome patients to try and identify cellular mechanisms that might underpin or correlate with specific clinical outcomes.
Hypothesis
In comparison with healthy controls, patients could have significant specific grey matter volume deficits and white matter hypo-connectivity; aberrant neuro-hemodynamic response involving frontal, striatal & limbic brain regions during fMRI; abnormal brain electrical activity and antisaccade/ smooth pursuit eye movements; prominent and specific patterns of impairments in verbal memory, verbal fluency, sustained attention and executive functions.
Unaffected relatives could also have brain abnormalities; and a proportion of these individuals may develop a clinical syndrome. A composite endophenotype comprising of neuroimaging and neurocognitive parameters could differentiate three groups (affected subjects, unaffected relatives & matched healthy controls). Subjects could show the deficits even in periods of clinical remission; and in a proportion of severely ill subjects these deficits may progress over a period.
Cellular assays could similarly differentiate cell lines derived from the three groups. Greater brain abnormalities/composite endophenotype measures/cellular abnormalities at baseline may predict poorer course and outcome/ treatment response. The relation between these, and the genetic variations, would be amenable for further analysis, to understand the genotype-phenotype conversion.
Clinical recruitment and work-plan
The affected probands will be recruited from the adult psychiatry services and specialty clinics (center of addiction medicine clinic, schizophrenia clinic, OCD clinic and geriatric clinic) of the NIMHANS, Bangalore. Unaffected FDRs will also be recruited from the families. An attempt will be made to recruit as many individuals as possible from each family. All clinics will together recruit age and gender matched control subjects, who will neither have an axis I psychiatric disorder, nor have a family history of such disorders in two previous generations.
All recruited individuals (N = 4500) will undergo brief clinical assessments and a blood sample will be drawn for isolation of DNA and peripheral blood mononuclear cells (PBMCs). From among this set, a subset of 1500 individuals (300 families and 300 controls) will be assessed at baseline on several additional assessments [detailed clinical assessments, endophenotype measures (neuroimaging, neuropsychology, psychophysics)], with the intention of conducting repeated measurements every alternate year. These individuals will form the neurodevelopmental endophenotype cohort (NEC). PBMCs from the NEC will be used to generate lymphoblastoid cell lines and a subset of this would be converted to IPS lines and also undego whole exome sequencing.
Specific methods
Clinical assessments
The study includes two levels of assessments - brief assessments and the NEC assessments. Brief, standardized assessments of overall health with a view to recording any pre-existing medical conditions or co-morbidity will be performed on all individuals who consent to participate in the study. This will provide diagnostics and psychometric evaluations that are compatible with international practice, and allow comparisons to be drawn. In addition, a more extensive endophenotype assessment will be performed on those families consenting to be part of the NEC. For the eligible subjects who consent for the endophenotype assessments, further detailed clinical evaluation of brain function will be done. This will include cross-disorder measures to assess temperament, personality, adverse childhood experiences, life events, handedness, socioeconomic status, functioning and psychopathology-specific scales. All clinical assessments that will form part of the assessment of the NEC are listed in Table 1.
Endophenotype assessments
The development of obvious clinical features in psychiatric diseases is usually preceded by a long period during which there are likely to be changes in brain function that contribute to disease development. We propose to map the development of these changes in the NEC using a number of complementary approaches. Previous research from NIMHANS and elsewhere have identified structural & functional brain imaging, eye movement parameters, electroencephalography (EEG) measures and cortical hemodynamic changes as robust endophenotypes [23, 26, 28,29,30,31,32,33,34,35,36,37,38,39,40,41,42] However, these assessments have largely been performed as single time measurements in patients with fully developed disease, their relatives and controls. These studies demonstrate that patients and their relatives differ from control subjects on these parameters. To capture the evolution of these important quantitative endophenotypes during critical stages of disease development, from the asymptomatic state, through to various stages of clinical disease, we propose to perform these measurements at two-year intervals in patients, unaffected FDR and healthy controls. The NEC assessments are outlined in Table 1.
Next-generation sequencing
Heritability estimates for severe mental illnesses range from 60% - 90%, suggesting that a large proportion of etiological variance is attributable to genetic factors [43,44,45]. Despite this evidence, additive effects of loci implicated by large scale genome-wide association studies (GWAS) have been estimated to explain only a small portion of this liability [46]. This has resulted in a shift in focus from common variants of small effect to rare and de-novo variants as determinants of disease causation. The rapid evolution of next generation sequencing platforms and falling costs of sequencing have enabled the identification of some of these rare and de-novo variants. Families with multiple affected members across generations are a critical resource as they have greater probability of segregating these putative causative variants for severe mental illness. The standard operating procedure (SOP) for next-generation sequencing in DBNS is given in Fig. 1. During this study, we propose to perform whole exome sequencing from affected individuals and their unaffected family members. The availability of such sequence along with suitable bioinformatics analysis will facilitate the identification of novel variants that may segregate with disease or specific aspects of illness such as responsiveness or resistance to specific treatment.

Whole exome sequencing
Cellular models: National Biorepository for mental illness
Critical to understanding the evolution of neuropsychiatric syndromes is the unravelling of cellular and sub-cellular processes that are altered in a given syndrome. This poses a unique challenge in the study of the brain related diseases, as the tissue is inaccessible. The availability of living brain cells is critical to study ongoing cellular processes in neurons and glial cells. Recent developments in stem cell technology allow the use of somatic tissues to be reprogrammed into IPS lines. These can then be differentiated into neural cells including neurons and glia. We propose to use this approach to derived cellular models of disease in the context of neuropsychiatric illness. Methods have been developed, that allow the establishment of stable cell lines from lymphocytes isolated from a sample of peripheral blood [47]. These LCLs can then be converted to IPS lines using non-integrating plasmids, which are free of transgene footprints and ideal for modelling disease mechanism [48, 49]. IPS lines can be differentiated into specific neuronal/glial cell types as well as composite cultures using a variety of available methods. Study of cellular and molecular processes in patient derived neuronal cell lines of a defined genetic background can be correlated with documented clinical semiology and the detailed endophenotype data. The SOPs for biomaterial processing is given in Fig. 2.

Biomaterial processing: Peripheral blood mononuclear cells (PBMNC) are extracted from blood. These cells are converted to lymphoblastoid cell lines (LCLs), which are then subsequently reprogrammed to generate human induced pluripotent stem cells (HiPSC). The HiPSCs are further transformed to neural stem cells (NSC). Quality control (QC) and characterization at each step is shown [49]
The functional unit of the brain is the synapse and it is likely that alterations in synapse formation, structure and function underlie neuropsychiatric illness. Synapse development and function is underpinned by complex sub-cellular processes and the “disease in a dish” model offers a unique opportunity to understand these events and their role in mental illness. To functionally characterize the IPS derived neural cell models we will test for neuronal activity. This will be done using whole cell patch-clamp recordings to test for the ability of neurons to generate action potentials. In addition, we will also measure function through the measurement and analysis of calcium transients using fluorescent calcium indicator dyes. More detailed analysis of response to neurotransmitters can also be performed using these approaches. In addition, neural networks will be examined in cultures using multi-electrode arrays.
Given the neurodevelopmental nature of mental illness, it is likely that altered synapse formation and pruning plays a role in disease development. To understand this process, we propose to characterize synapse development in our cellular models. This can be done by differentiating IPS into neurons in cell culture and characterizing neurite outgrowth during this process using well-established quantitative assays.
Metabolic alterations in neural cells are strongly implicated in mental illness and multiple roles for mitochondrial dysfunction in the development of several mental disorders is widely discussed. To map these potential defects, we will perform basic characterization of mitochondrial structure and function using well-established fluorescence based assays. Metabolic alterations arising from altered mitochondrial function or additional genetic factors will be captured by metabolic profiling using small molecule mass-spectrometry. Such analyses may also include changes in the levels of neurotransmitters.
Sample size calculation and statistical analysis
The sample size was calculated for each trait and number of times of assessment separately for each trait [50] for repeated measurements. The required number of families (n) assuming independence of subjects, can be obtained using [50], as follows.
Let σ2 is the assumed common variance in the two groups, (μ1 − μ2) is the difference in means of the two groups, k is the number of time points and ρ is the assumed correlation of the repeated measures; then the number of subjects/ families (assuming independence) in each of two groups is,
The power and type I error were kept at 80% and 5% respectively.
If the affected group and high-risk group (FDRs) have an average family size of ‘m’, thereby the total number of observations = ‘nm’, then the effective sample would be N = nm/[1 + (m-1) λ]; where ‘λ’ is the intra-familial correlation (intra-class correlation) on the quantitative trait.
We have used this N in the standard power computation formula for unbalanced ANOVA for the number of observations in the affected group and FDRs along with the control group.
Using this formula, we have calculated sample size using brain imaging and brain physiology measures. These are as follows:
-
a.
Corpus callosal volume: This white matter tract has been implicated in all major psychiatric disorders and has also been examined in large scale studies. Using published literature on corpus callosal volume [51], sample size calculation as per above formula reveals 1509 participants overall for 80% power.
-
b.
Eye movement parameters: Eye movement abnormalities have been identified as putative endophenotypes in recent large studies [52]. Sample size calculated for antisaccade latency was 1257 participants overall for 80% power.
These power calculations described above will adequately address our capacity to detect group differences. Moreover, as we detect genetic differences (rare variants/ accumulation of damaging variants), and also submit the genetic data generated to pathway based modelling, these emerging data will be used to analyse MRI and EEG data in a specific manner.
Discussion
Longitudinal assessment of biological measures
The human brain is a complex organ and its output, i.e. manifest behavior, arises from processes operating at multiple scales ranging from individual molecules within cells and integrating into increasingly higher levels of complexity including cell- cell interactions and circuit formation leading to emerging structure and function. These multi-scale events are underpinned not only by genetic factors that determine the functional biochemistry of brain cells, but also by epigenetic factors that modulate the expression of genes in an individual genome. Given the inherent variability between the genome of individuals as well as the range of environmental factors that can influence gene expression, it is not surprising that uncovering the etiology and pathogenesis of mental illness remains one of the greatest challenges in modern biology. To meet this challenge, our proposal to assemble a cohort of clinically dense families and prospectively follow the development of disease in them, would be useful. The use of clinically dense families coupled with next generation sequencing offers an opportunity to uncover rare and de novo variants and study their segregation pattern within a family. These observations could offer insights into the inherited basis of mental illness, as also be extrapolated to the whole population. Simultaneously, the detailed clinical history and demographics collected for members of the cohort will offer an insight into environmental factors that may mediate epigenetic regulation of the genome in an individual and therefore explain the development of mental illness [53].
Numerous case-control studies have identified changes in brain structure and function at multiple levels. However, we are unaware of any study which has mapped the development of such changes from at-risk state to disease onset, using multiple measures. Most of our knowledge of brain perturbations that lead to psychopathology has been derived from cross-sectional “snap-shots”, or explorations which commence after the development of a recognizable syndromal state. The specific roles that genes, environment and their interaction play in typical and atypical development are unclear, especially across the course of mental illness. Existing knowledge of brain and behavioral development is insufficient to permit identification of specific time points at which normal and psychopathological trajectories diverge and the processes that underlie such deviations. We propose to set up a prospective longitudinal cohort of individuals who are at-risk of developing psychiatric disorders and follow them with periodic endophenotype assessment over time. This approach offers the opportunity to map structural and functional changes in brain function as they develop, along with behavioral changes that lead to disease development. This is likely to be vastly more informative than previous cross-sectional case-control studies. We will use advanced imaging methods to accurately track, changes in brain structure and connectivity (both structural and functional), the neurodevelopmental processes related to the normal developmental sequence and deviations from it. In contrast to conventional genetic association studies where disease status is based on clinical observations, neuroimaging phenotypes can capture aspects of disease phenotypes at the physiological level. Neuroimaging genetic studies thus offer the prospect of gains in statistical power, since genes code not for mental or behavioural traits, but for the neural phenotypes that underpin them [54]. Neuroimaging genetic studies have the additional benefit of spatially localizing gene effects, offering further potential insights into the structural and functional neurobiology of disease [55,56,57,58]. Thus, the availability of this temporal data set may offer predictive value in relating specific changes in brain structure and function to the development of clinical features or response to treatment. Uncovering such changes may offer clinical psychiatry the opportunity to personalize treatment; e.g. when a specific early change in brain structure or function predicts a clinical outcome. They may also offer the opportunity to predict treatment response and therefore offer personalized medicine.
Aggregation of syndromes with varied symptomatology
The validity of the discrete disease constructs (constructed mainly based on phenomenological similarities and dissimilarities) and classificatory models of mental disorders, which have hitherto been adequate for clinical purposes, have been strongly challenged by recent findings of wide overlaps between these discrete entities in terms of genetic underpinnings, neurodevelopmental substrates, responses to pharmacological agents and behavioural treatments [9,10,11,12,13,14,15]. In fact, considerable overlap is present in the phenomenology as well [9, 59]. Moreover, these disorders do not necessarily breed true and it is not uncommon to find that an index case may have a family history of a different psychiatric illness [59,60,61]. This lack of validity of a symptom based classification is considered as one of the main reasons for the absence of diagnostic specificity of biological observations [62]. It is likely that the recognizable, syndromal disease states are due to deviations on the central trajectory of the normal brain developmental sequence which includes processes like neuronal migration, proliferation, pruning and myelination.
By recruiting individuals from different disorders and mapping endophenotypes across different domains, we hope that we will be able to achieve increased power to identify the common neurobiological factors underlying severe mental illness. Such attempts have been made by recent research consortia like the Bipolar-Schizophrenia Network for Intermediate Phenotypes (B-SNIP) and Consortium on the Genetics of Schizophrenia (COGS) [10, 11, 63]. While these initiatives were restricted to focused on the psychosis spectrum, in the present study we aim to investigate beyond psychosis spectrum; and additionally, use cellular models.
Of the neuropsychiatric disorders that we have included in this study, the outlier is dementia, which usually manifests at a later age than the others. However, dementia is also increasingly recognized to have its beginnings in adolescence, as early studies point to recognizable brain changes in persons at high risk, starting many years before the recognizable symptoms of the disease are palpable [64,65,66,67]. Moreover, overlaps in symptoms, risk to family members and genetic factors are evident [68, 69]. Such findings support the inclusion of AD patients in a cohort of neurodevelopmental disorders.
Combination of clinical endophenotypes and IPS
It is now possible to assemble and manipulate so-called neural spheroids, generated from human induced pluripotent stem cells, to study the normal development of the human brain and deviations from the developmental sequence. Since many neuropsychiatric disorders are possibly influenced by individual genetic influences, it is difficult to study these diseases in standard animal models. Instead, these types of diseases can be modeled using the cells from the patient. One can use the information from longitudinal tracking of the basic processes underlying brains fundamental neurodevelopmental sequence in normal development and in disease, to a] investigate the internal cellular processes, underlying the processes involved in neurodevelopment, b] Model, deviations in cellular processes, which lead to deviations in neurodevelopmental processes, and c] test, the processes and outcomes of physical and drug treatments.
Resources to be generated and expected outcomes
The unified digital database of clinical-endophenotype measures and the bio-repository generated in DBNS will provide a wealth of quantitative detail, which can then be interrogated both within and across phenotype (clinical overlaps/outcome) as well as genetic data (presence of at-risk alleles, or sharing). Other than the specified objectives, some of the other expected off-shoots of the project are as follows:
Genetic basis of psychiatric endophenotypes
The program seeks to identify valid endophenotypes in psychiatry using a large sample; and have next generation sequencing data for the same set of individuals. Using appropriate statistical techniques for data reduction (e.g: principal components analysis) and endophenotype-genetics relationships (e.g: parallel independent component analysis), we hope to identify the genetic basis of these endophenotypes. Similar endophenotype-genetics analyses (n = 1250) have been recently published from the B-SNIP cohort [37, 70].
Mechanisms/predictors of drug response/adverse events
Availability of clinical-endophenotype measures and biomaterial from individuals who have/haven’t responded to particular drugs/ have adverse events to specific drugs will be valuable tools in this approach. Cellular models from such individuals can also be used for testing drug effects in-vitro and for the identification of new drug targets [71]. Such an approach has already been used for Lithium response in bipolar disorder [72] and needs to be extended to other disorders and drugs.
Gene editing experiments
We hope to identify damaging rare variants in the loaded families using next generation sequencing. Advances in genome engineering techniques based on the CRISPR-associated RNA-guided endonuclease Cas9 allow us to create isogenic cell lines, either by inserting these mutations into a control cell line, or by correction of the mutations in a cell line from a diseased individual. The advantage of such an approach is that each modified cell line will have its own control, the only difference being the edited mutation with the genomic background unaltered.
Combination of cellular models
Many of the biological signals being identified in psychiatry indicate involvement of systemic genes. The use of IPS derived neuronal lineage cells alone may not capture these systemic effects; hence, a combination of IPS derived neurons, glia, and peripheral model systems (e.g., LCLs, lymphocytes) will prove useful. Additional use of peripheral cells will also advance bench to bedside translation.
Biological predictors of disease conversion
The unique feature of this program is the combination of a clinical longitudinal approach with prospective assessment of biological measures. A proportion of the FDRs will convert to clinical illness during the follow-up period. The program should be able to identify measurable predictors of disease conversion in FDRs at various biological levels – brain imaging, psycho-physics, epigenetics etc. In addition, examination of cellular endophenotypes across FDRs who have converted to clinical disease and who have not, in the same family, may point towards disease conversion related functional output at the cellular level.
Conclusion
The DBNS is thus an ambitious, and optimistic, attempt to create a database that combines a wealth of clinical data with a comprehensive psychological and biological assessment, over time. The resources generated will hopefully serve as a platform to answer several questions related to the neurobiology of psychiatric disorders; as well as address fundamental questions about neuro-development and degeneration, as relevant to common psychiatric disease syndromes. This would improve our understanding of these illnesses, and perhaps develop strategies for amelioration, of these conditions which are a significant public health concern.
Abbreviations
- AD:
-
Alzheimer’s dementia
- ADBS:
-
Accelerator program for discovery in brain disorders using stem cells
- BD:
-
Bipolar disorder
- DBNS:
-
Discovery biology of neuropsychiatric syndromes
- EEG:
-
Electroencephalography
- FDR:
-
First degree relative
- GWAS:
-
Genome-wide association study
- IPS:
-
Induced pluripotent stem cell
- LCL:
-
Lymphoblastoid cell lines
- MRI:
-
Magnetic resonance imaging
- NEC:
-
Neurodevelopmental endophenotype cohort
- OCD:
-
Obsessive compulsive disorder
- PBMC:
-
Peripheral blood mononuclear cell
- SOP:
-
Standard operating procedure
- SUD:
-
Substance use disorder
References
WHO. The world health report 2001: mental health: new understanding, new hope. World Health Organization; 2001.
Kessler RC, Amminger GP, Aguilar-Gaxiola S, Alonso J, Lee S, Ustun TB. Age of onset of mental disorders: a review of recent literature. Curr Opin Psychiatry. 2007 ;20:359–364. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17551351 [cited 2017 Apr 14].
Waltereit R, Banaschewski T, Meyer-Lindenberg A, Poustka L. Interaction of neurodevelopmental pathways and synaptic plasticity in mental retardation, autism spectrum disorder and schizophrenia: implications for psychiatry. World J Biol Psychiatry. 2014;15:507–516. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24079538 [cited 2017 Jul 10].
Giegling I, Hosak L, Mössner R, Serretti A, Bellivier F, Claes S, et al. Genetics of schizophrenia: A consensus paper of the WFSBP Task Force on Genetics. World J. Biol. Psychiatry. 2017 ;1–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28112043 [cited 2017 Jul 10].
McGorry PD, Purcell R, Goldstone S, Amminger GP. Age of onset and timing of treatment for mental and substance use disorders: implications for preventive intervention strategies and models of care. Curr. Opin. Psychiatry. 2011;24:301–306. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21532481 [cited 2017 Apr 14].
Zúñiga A, Navarro JB, Lago P, Olivas F, Muray E, Crespo M. Evaluation of needs among patients with severe mental illness. A community study. Actas Esp Psiquiatr. ;41:115–121. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23592071 [cited 2017 Jul 10].
Swain S, Behura S. A comparative study of quality of life and disability among schizophrenia and obsessive-compulsive disorder patients in remission. Ind Psychiatry J. 2016;25:210. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28659702 [cited 2017 Jul 10].
Shih R a, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatry. 2004;16:260–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16194760.
Craddock N, Owen MJ. The Kraepelinian dichotomy - going, going... But still not gone. Br J Psychiatry Royal College of Psychiatrists; 2010;196:92–95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20118450 [cited 2017 Apr 14].
Ethridge LE, Hamm JP, Shapiro JR, Summerfelt AT, Keedy SK, Stevens MC, et al. Neural activations during auditory oddball processing discriminating schizophrenia and psychotic bipolar disorder. Biol. Psychiatry. 2012 ;72:766–774. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22572033 [cited 2017 Apr 14].
Meda SA, Gill A, Stevens MC, Lorenzoni RP, Glahn DC, Calhoun VD, et al. Differences in resting-state functional magnetic resonance imaging functional network connectivity between schizophrenia and psychotic bipolar Probands and their unaffected first-degree relatives. Biol. Psychiatry. 2012 ;71:881–889. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22401986 [cited 2017 Apr 14].
Docherty AR, Moscati AA, Fanous AH. Cross-disorder psychiatric genomics. Curr Behav Neurosci Reports 2016 [cited 2017 Jul 10];3:256–263. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28042526.
McTeague LM, Huemer J, Carreon DM, Jiang Y, Eickhoff SB, Etkin A. Identification of common neural circuit disruptions in cognitive control across psychiatric disorders. Am. J. Psychiatry. 2017 ;174:676–685. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28320224 [cited 2017 Jul 25].
McTeague LM, Goodkind MS, Etkin A. Transdiagnostic impairment of cognitive control in mental illness. J Psychiatr Res. 2016 ;83:37–46. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0022395616301868 [cited 2017 Jul 25].
Buckholtz JW, Meyer-Lindenberg A. Psychopathology and the human connectome: toward a Transdiagnostic model of risk for mental illness. Neuron. 2012 ;74:990–1004. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0896627312005144 [cited 2017 Jul 25].
Lee SH, Ripke S, Neale BM, Faraone S V, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet.. 2013 ;45:984–994. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23933821 [cited 2017 Jun 10].
Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013 ;381:1371–1379. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23453885 [cited 2017 Jun 10].
Gonzalez-Mantilla AJ, Moreno-De-Luca A, Ledbetter DH, Martin CL. A cross-disorder method to identify novel candidate genes for developmental brain disorders. JAMA Psychiatry. 2016 ;73:275. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26817790 [cited 2017 Jun 10].
Cicchetti D, Cannon TD. Neurodevelopmental processes in the ontogenesis and epigenesis of psychopathology. Dev Psychopathol. 1999 ;11:375–393. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10532615 [cited 2017 Apr 14].
Sanjay TN, Shivakumar V, Subbanna M, Biradar SU, Muralidharan K, Venkatasubramanian G. Plasma interleukin-6 in remitted early bipolar I disorder and subjects at high-risk for bipolar disorder. Asian J. Psychiatr.. 2017 ; Available from: http://www.ncbi.nlm.nih.gov/pubmed/28365164 [cited 2017 Jul 10].
Kulkarni S, Jain S, Janardhan Reddy Y, Kumar KJ, Kandavel T. Impairment of verbal learning and memory and executive function in unaffected siblings of probands with bipolar disorder. Bipolar Disord. 2010 ;12:647–656. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20868463 [cited 2017 Jan 18].
Harave V, Shivakumar V, Kalmady S, Narayanaswamy J, Varambally S, Venkatasubramanian G. Neurocognitive impairments in unaffected first-degree relatives of schizophrenia. Indian J Psychol Med. 2017 ;39:250. Available from: http://www.ijpm.info/text.asp?2017/39/3/250/207335 [cited 2017 Jul 10].
Benegal V, Antony G, Venkatasubramanian G, Jayakumar PN. Gray matter volume abnormalities and externalizing symptoms in subjects at high risk for alcohol dependence. Addict Biol. 2007 ;12:122–132. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17407506 [cited 2017 Apr 24].
Muralidharan K, Venkatasubramanian G, Pal PK, Benegal V. Relationship between motor threshold and externalizing symptoms in subjects at high risk for alcohol dependence: a TMS study. Am J Addict. 2013 ;22:84–85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23398231 [cited 2017 Jul 10].
Holla B, Panda R, Venkatasubramanian G, Biswal B, Bharath RD, Benegal V. Disrupted resting brain graph measures in individuals at high risk for alcoholism. Psychiatry Res Neuroimaging. 2017 ;265:54–64. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28531764 [cited 2017 Jul 10].
Viswanath B, Janardhan Reddy YC, Kumar KJ, Kandavel T, Chandrashekar CR. Cognitive endophenotypes in OCD: a study of unaffected siblings of probands with familial OCD. Prog Neuro-Psychopharmacology Biol Psychiatry. 2009 ;33:610–615. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19272409 [cited 2017 Apr 14].
Dean DC, Jerskey BA, Chen K, Protas H, Thiyyagura P, Roontiva A, et al. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. JAMA Neurol. 2014 ;71:11–22. Available from: http://archneur.jamanetwork.com/article.aspx?doi=10.1001/jamaneurol.2013.4544 [cited 2017 Jul 10].
Muralidharan K, Venkatasubramanian G, Pal PK, Benegal V. Abnormalities in cortical and transcallosal inhibitory mechanisms in subjects at high risk for alcohol dependence: a TMS study. Addict. Biol.. 2008 ;13:373–379. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18422835 [cited 2017 Apr 24].
John JP, Khanna S, Pradhan N, Mukundan CR. EEG alpha coherence and psychopathological dimensions of schizophrenia. Indian J Psychiatry. 2002 ;44:97–107. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21206554 [cited 2017 Apr 14].
John JP, Rangaswamy M, Thennarasu K, Khanna S, Nagaraj RB, Mukundan CR, et al. EEG power spectra differentiate positive and negative subgroups in neuroleptic-naive schizophrenia patients. J Neuropsychiatry Clin Neurosci. 2009 ;21:160–172. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19622687 [cited 2017 Apr 14].
Behere RV, Raghunandan V, Venkatasubramanian G, Subbakrishna DK, Jayakumar PN, Gangadhar BN. Trends-a tool for recognition of emotions in neuropsychiatric disorders. Indian. J Psychol Med Medknow Publications. 2008;30:32.
Venkatasubramanian G, Puthumana DTK, Jayakumar PN, Gangadhar BN. A functional magnetic resonance imaging study of neurohemodynamic abnormalities during emotion processing in subjects at high risk for schizophrenia. Indian J. Psychiatry. Medknow Publications; 2010 ;52:308–315. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21267363 [cited 2017 Apr 14].
John JP, Halahalli HN, Vasudev MK, Jayakumar PN, Jain S. Regional brain activation/deactivation during word generation in schizophrenia: fMRI study. Br. J. Psychiatry. 2011 ;198:213–222. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21357880 [cited 2017 Apr 14].
KOLUR US, REDDY YCJ, JOHN JP, KANDAVEL T, JAIN S. Sustained attention and executive functions in euthymic young people with bipolar disorder. Br J Psychiatry. 2006 ;189:453–458. Available from: http://bjp.rcpsych.org/cgi/doi/10.1192/bjp.bp.106.022921 [cited 2017 Apr 14].
Narayanan B, O’Neil K, Berwise C, Stevens MC, Calhoun VD, Clementz BA, et al. Resting state electroencephalogram oscillatory abnormalities in schizophrenia and psychotic bipolar patients and their relatives from the bipolar and schizophrenia network on intermediate phenotypes study. Biol Psychiatry. 2014 ;76:456–465. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24439302 [cited 2017 Apr 14].
Narayanan B, Soh P, Calhoun VD, Ruaño G, Kocherla M, Windemuth A, et al. Multivariate genetic determinants of EEG oscillations in schizophrenia and psychotic bipolar disorder from the BSNIP study. Transl Psychiatry. 2015 ;5:e588. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26101851 [cited 2017 Apr 14].
Meda SA, Ruano G, Windemuth A, O’Neil K, Berwise C, Dunn SM, et al. Multivariate analysis reveals genetic associations of the resting default mode network in psychotic bipolar disorder and schizophrenia. Proc Natl Acad Sci. 2014 ;111:E2066–E2075. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24778245 [cited 2017 Apr 14].
Hibar DP, Westlye LT, van Erp TGM, Rasmussen J, Leonardo CD, Faskowitz J, et al. Subcortical volumetric abnormalities in bipolar disorder. Mol Psychiatry. 2016 ;21:1710–1716. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26857596 [cited 2017 Apr 14].
Boedhoe PSW, Schmaal L, Abe Y, Ameis SH, Arnold PD, Batistuzzo MC, et al. Distinct subcortical volume alterations in pediatric and adult OCD: a worldwide Meta- and mega-analysis. Am J Psychiatry. 2017 ;174:60–69. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27609241 [cited 2017 Apr 14].
Silva MC, Benegal V, Devi M, Mukundan CR. Cognitive deficits in children of alcoholics: at risk before the first sip! Indian J Psychiatry Medknow Publications; 2007 ;49:182–188. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20661384 [cited 2017 Apr 24].
Benegal V, Jain S, Subbukrishna DK, Channabasavanna SM. P300 amplitudes vary inversely with continuum of risk in first degree male relatives of alcoholics. Psychiatr Genet. 1995 ;5:149–156. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8750356 [cited 2017 Apr 24].
Shaikh KJ, Naveen D, Sherrin T, Murthy A, Thennarasu K, Anand A, et al. Polymorphisms at the DRD2 locus in early-onset alcohol dependence in the Indian population. Addict. Biol.. 2001 ;6:331–335. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11900611 [cited 2017 Apr 24].
McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry. 2003;60:497–502.
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait. Arch Gen Psychiatry. 2003 ;60:1187. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14662550 [cited 2017 Apr 14].
Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012 ;13:537–551. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22777127 [cited 2017 Apr 14].
Lee SH, Ripke S, Neale BM, Faraone S V, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013 ;45:984–994. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23933821[cited 2017 Apr 14].
Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986 ;73:320–326. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3017841 [cited 2015 Oct 4].
Shirley MD, Baugher JD, Stevens EL, Tang Z, Gerry N, Beiswanger CM, et al. Chromosomal variation in lymphoblastoid cell lines. Hum Mutat. 2012 ;33:1075–1086. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22374857 [cited 2017 Apr 14].
Muthusamy T, Mukherjee O, Menon R, Megha PB, Panicker MM. A method to identify and isolate pluripotent human stem cells and mouse epiblast stem cells using lipid body-associated retinyl ester fluorescence. Stem cell reports Elsevier; 2014 ;3:169–184. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25068130 [cited 2017 Apr 14].
Diggle P, Heagarty P, Liang KY, Zeger S. Analysis of longitudinal data. Oxford University Press. 2002;ISBN 0198524846.
Francis AN, Mothi SS, Mathew IT, Tandon N, Clementz B, Pearlson GD, et al. Callosal abnormalities across the psychosis dimension: bipolar schizophrenia network on intermediate phenotypes. Biol. Psychiatry. 2016 ;80:627–635. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26954565 [cited 2017 Apr 14].
Lencer R, Sprenger A, Reilly JL, McDowell JE, Rubin LH, Badner JA, et al. Pursuit eye movements as an intermediate phenotype across psychotic disorders: evidence from the B-SNIP study. Schizophr. Res NIH Public Access; 2015 ;169:326–333. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26481615 [cited 2017 Jan 18].
Lahiri DK, Maloney B, Bayon BL, Chopra N, White FA, Greig NH, et al. Transgenerational latent early-life associated regulation unites environment and genetics across generations. Epigenomics. 2016 ;8:373–387. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26950428 [cited 2017 Jul 17].
Glahn DC, Thompson PM, Blangero J. Neuroimaging endophenotypes: strategies for finding genes influencing brain structure and function. Hum Brain Mapp. 2007 ;28:488–501. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17440953 [cited 2017 Jun 10].
Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006 ;7:818–827. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16988657 [cited 2017 Apr 14].
Roffman JL, Weiss AP, Goff DC, Rauch SL, Weinberger DR. Neuroimaging-genetic paradigms: a new approach to investigate the pathophysiology and treatment of cognitive deficits in schizophrenia. Harv Rev Psychiatry. 2006 ;14:78–91. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16603474 [cited 2017 Jun 10].
Hariri AR, Drabant EM, Weinberger DR. Imaging genetics: perspectives from studies of genetically driven variation in serotonin function and Corticolimbic affective processing. Biol. Psychiatry. 2006 ;59:888–897. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16442081 [cited 2017 Jun 10].
Bogdan R, Salmeron BJ, Carey CE, Agrawal A, Calhoun VD, Garavan H, et al. Imaging genetics and genomics in psychiatry: a critical review of progress and potential. Biol. Psychiatry. 2017 ;82:165–175. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28283186 [cited 2017 Jul 10].
Ivleva EI, Morris DW, Moates AF, Suppes T, Thaker GK, Tamminga CA. Genetics and intermediate phenotypes of the schizophrenia—bipolar disorder boundary. Neurosci Biobehav Rev. 2010 ;34:897–921. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19954751 [cited 2017 Apr 14].
Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009 ;373:234–239. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19150704 [cited 2017 Apr 14].
Narayanaswamy JC, Varghese M, Jain S, Sivakumar PT, Prakash O, Bharath S, et al. Is there a familial overlap between dementia and other psychiatric disorders? Int Psychogeriatrics. 2011 ;23:749–755. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21205363 [cited 2017 Jul 10].
Keshavan MS. Nosology of psychoses in DSM-5: Inches ahead but miles to go. Schizophr. Res. 2013. p. 40–1. Available from: http://www.sciencedirect.com/science/article/pii/S0920996413003769 [cited 2017 Apr 14].
Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ, et al. Analysis of 94 candidate genes and 12 Endophenotypes for schizophrenia from the consortium on the genetics of schizophrenia. Am. J. Psychiatry. 2011 ;168:930–946. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21498463 [cited 2017 Apr 14].
Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, et al. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurol. 2007 ;6:494–500. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17509484 [cited 2017 Jun 10].
Chang L, Douet V, Bloss C, Lee K, Pritchett A, Jernigan TL, et al. Gray matter maeturation and cognition in children with different APOE ε genotypes. Neurology. 2016 ;87:https://doi.org/10.1212/WNL.0000000000002939. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27412137 [cited 2017 Jul 10].
Calderón-Garcidueñas L, Mora-Tiscareño A, Melo-Sánchez G, Rodríguez-Díaz J, Torres-Jardón R, Styner M, et al. A critical proton MR spectroscopy marker of Alzheimer’s disease early neurodegenerative change: low hippocampal NAA/Cr ratio impacts APOE ɛ4 Mexico City children and their parents. J Alzheimers Dis. 2015 ;48:1065–1075. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26402110 [cited 2017 Jul 10].
Dean DC, Jerskey BA, Chen K, Protas H, Thiyyagura P, Roontiva A, et al. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease. JAMA Neurol.. 2014 ;71:11. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24276092 [cited 2017 Jul 10].
DeMichele-Sweet MAA, Weamer EA, Klei L, Vrana DT, Hollingshead DJ, Seltman HJ, et al. Genetic risk for schizophrenia and psychosis in Alzheimer disease. Mol. Psychiatry. 2017 ; Available from: http://www.ncbi.nlm.nih.gov/pubmed/28461698 [cited 2017 Jul 17].
Liebers DT, Pirooznia M, Seiffudin F, Musliner KL, Zandi PP, Goes FS. Polygenic risk of schizophrenia and cognition in a population-based survey of older adults. Schizophr Bull. 2016 ;42:984–991. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26873889 [cited 2017 Jul 17].
Narayanan B, Ethridge LE, O’Neil K, Dunn S, Mathew I, Tandon N, et al. Genetic sources of subcomponents of event-related potential in the dimension of psychosis analyzed from the B-SNIP study. Am. J. Psychiatry. 2015 ;172:466–478. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25615564 [cited 2017 Apr 14].
Morag A, Kirchheiner J, Rehavi M, Gurwitz D. Human lymphoblastoid cell line panels: novel tools for assessing shared drug pathways. Pharmacogenomics. 2010 ;11:327–340. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20235789 [cited 2017 Apr 14].
Mertens J, Wang Q-W, Kim Y, Yu DX, Pham S, Yang B, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015 ;527:95–99. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26524527 [cited 2017 Apr 14].
Narrow WE, Clarke DE, Kuramoto SJ, Kraemer HC, Kupfer DJ, Greiner L, et al. DSM-5 field trials in the United States and Canada, part III: development and reliability testing of a cross-cutting symptom assessment for DSM-5. Am. J. Psychiatry. 2013 ;170:71–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23111499 [cited 2017 Apr 14].
Sheehan D V, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The mini-international neuropsychiatric interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998 ;59 Suppl 2:2233–2257. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9881538 [cited 2014 Jul 10].
Maxwell ME. Family interview for genetic studies (FIGS): a manual for FIGS. Clin. Neurogenetics branch, intramural res. Bethesda, MD: Program, Natl. Inst. Ment. Heal; 1992.
Adler LA, Kessler RC, Spencer T. Adult ADHD self-report scale-v1. 1 (ASRS-v1. 1) symptom checklist. New York, NY: World Heal. Organ; 2003.
Ganguli M, Ratcliff G, Chandra V, Sharma S, Gilby J, Pandav R, et al. A Hindi version of the MMSE: the development of a cognitive screening instrument for a largely illiterate rural elderly population in India. Int J Geriatr Psychiatry Wiley Online Library. 1995;10:367–77.
Guy W. ECDEU assessment manual for psychopharmacology. US Department of Health, education, and welfare, public health service, alcohol, drug abuse, and mental health administration, National Institute of Mental Health, psychopharmacology research branch, division of extramural research programs; 1976.
Evans DE, Rothbart MK. Developing a model for adult temperament. J Res Pers. 2007 ;41:868–888. Available from: http://www.sciencedirect.com/science/article/pii/S0092656606001334 [cited 2017 Apr 14].
Donnellan MB, Oswald FL, Baird BM, Lucas RE. The mini-IPIP scales: tiny-yet-effective measures of the big five factors of personality. Psychol Assess. 2006 ;18:192–203. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16768595 [cited 2017 Apr 14].
Spitzer RL, Gibbon M, Williams JBW, Endicott J. Global assessment of functioning (GAF) scale. Outcome assess. Clin. Pract. Baltimore: Williams and Wilkins; 1996. p. 76–8.
Mundt JC, Marks IM, Shear MK, Greist JM. The work and social adjustment scale: a simple measure of impairment in functioning. Br J Psychiatry RCP. 2002;180:461–4.
Organization WH. Adverse childhood experiences international questionnaire (ACE-IQ). 2015.
Paykel ES. The interview for recent life events. Psychol Med Cambridge Univ Press. 1997;27:301–10.
Kuppuswamy B. Manual of socio-economic status scale. Delhi Manasayan Publ. 1962;.
Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia Elsevier. 1971;9:97–113.
Andreasen NC. Scale for the assessment of positive symptoms (SAPS). Iowa City: University of Iowa; 1984.
Andreasen NC. The Scale for the Assessment of Negative Symptoms (SANS): conceptual and theoretical foundations. Br. J. Psychiatry. Suppl.. 1989 ;49–58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2695141 [cited 2017 Apr 14].
Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. Br. J. Psychiatry. 1978 ;133:429–435. Available from: http://www.ncbi.nlm.nih.gov/pubmed/728692 [cited 2017 Apr 14].
HAMILTON M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960 ;23:56–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14399272 [cited 2017 Apr 14].
HAMILTON M. The assessment of anxiety states by rating. Br J Med Psychol. 1959 ;32:50–55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/13638508 [cited 2017 Apr 14].
Goodman WK, Price LH, Rasmussen SA, Mazure C, Fleischmann RL, Hill CL, et al. The Yale-Brown obsessive compulsive scale. I. Development, use, and reliability. Arch. Gen. Psychiatry. 1989 ;46:1006–1011. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2684084 [cited 2017 Apr 14].
Hughes CP, Berg L, Danziger WL, Coben LA, Martin R. A new clinical scale for the staging of dementia. Br J psychiatry RCP. 1982;140:566–72.
Strub RL, Black FW. The mental status examination in neurology. 3rd ed. FA Davis Company. 1993;ISBN 0-8036-8212-3.
D’Elia L, Satz P, Uchiyama CL, White T. Color trails test: professional manual. Odessa, FL: Psychological Assessment Resources; 1996.
KIRCHNER WK. Age differences in short-term retention of rapidly changing information. J Exp Psychol. 1958 ;55:352–358. Available from: http://www.ncbi.nlm.nih.gov/pubmed/13539317 [cited 2017 Apr 14].
Logan GD. On the ability to inhibit thought and action: a users’ guide to the stop signal paradigm. In Dagenbach D, Carr TH (Eds.), Inhibitory processes in attention, memory, and language. San Diego: Academic Press. 1994;189–239.
Rey A. L’examen psychologique dans les cas d’encéphalopathie traumatique.(Les problems.). Arch. Psychol. Geneve: Editions Médecine et Hygiène; 1941.
Mehta UM, Thirthalli J, Naveen Kumar C, Mahadevaiah M, Rao K, Subbakrishna DK, et al. Validation of social cognition rating tools in Indian setting (SOCRATIS): a new test-battery to assess social cognition. Asian J Psychiatr. 2011 ;4:203–209. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23051118 [cited 2017 Apr 14].
Bharadwaj B, Arasappa R, Behere R, Venkatasubramanian G, Jayakumar P, Gangadhar B. Emotion recognition deficits in antipsychotic-naive schizophrenia. Indian J Psychol Med Medknow Publications; 2008 ;30:90. Available from: http://www.ijpm.info/text.asp?2008/30/2/90/48481 [cited 2017 Apr 14].
Taylor AJG, Hutton SB. The effects of task instructions on pro and antisaccade performance. Exp Brain Res. 2009 ;195:5–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19283372 [cited 2017 Apr 14].
Hong LE, Turano KA, O’Neill H, Hao L, Wonodi I, McMahon RP, et al. Refining the predictive pursuit Endophenotype in schizophrenia. Biol. Psychiatry. 2008 ;63:458–464. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17662963 [cited 2017 Apr 14].
Ruocco AC, Rodrigo AH, Lam J, Di Domenico SI, Graves B, Ayaz H. A problem-solving task specialized for functional neuroimaging: validation of the Scarborough adaptation of the tower of London (S-TOL) using near-infrared spectroscopy. Front Hum Neurosci. 2014 ;8:185. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24734017 [cited 2017 Apr 14].
Santos JL, Sánchez-Morla EM, Aparicio A, García-Jiménez MÁ, Villanueva C, Martínez-Vizcaíno V, et al. P50 gating in deficit and nondeficit schizophrenia. Schizophr Res. 2010 ;119:183–190. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20153607 [cited 2017 Apr 14].
Acknowledgements
Biju Viswanath, Naren P. Rao, Janardhanan C. Narayanaswamy, Palanimuthu T. Sivakumar, Arun Kandasamy, Muralidharan Kesavan, Urvakhsh Meherwan Mehta, Ganesan Venkatasubramanian, John P. John, Odity Mukherjee, Meera Purushottam, Ramakrishnan Kannan, Bhupesh Mehta, Thennarasu Kandavel, B. Binukumar, Jitender Saini, Deepak Jayarajan, A. Shyamsundar, Sydney Moirangthem, K. G. Vijay Kumar, Jagadisha Thirthalli, Prabha S. Chandra, Bangalore N. Gangadhar, Pratima Murthy, Mitradas M. Panicker, Upinder S. Bhalla, Sumantra Chattarji, Vivek Benegal, Mathew Varghese, Janardhan Y. C. Reddy, Padinjat Raghu, Mahendra Rao and Sanjeev Jain.
Funding
This research is funded by the Accelerator program for discovery in brain disorders using stem cells (ADBS) (jointly funded by the Department of Biotechnology, Government of India, and the Pratiksha trust). BV is partially supported by the INSPIRE fellowship of the Department of Science and Technology, Government of India. GV is partially supported by the Swarnajayanti Fellowship of the Department of Science and Technology, Government of India. RK is funded by the Ramalingaswamy re-entry fellowship of the Department of Biotechnology, Government of India.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
This is a consortium paper and there are no individual authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study has been approved by the institutional ethics committee at all three participating institutes [(National Institute of Mental Health and Neuro Sciences (NIMHANS), the National Center for Biological Sciences (NCBS), and the Institute for Stem Cell Biology and Regenerative Medicine (InStem)]. A written informed consent will be obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Viswanath, B., Rao, N.P., Narayanaswamy, J.C. et al. Discovery biology of neuropsychiatric syndromes (DBNS): a center for integrating clinical medicine and basic science. BMC Psychiatry 18, 106 (2018). https://doi.org/10.1186/s12888-018-1674-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12888-018-1674-2