
Abstract
Background
Early-stage non-small cell lung carcinoma (NSCLC) accounts for more than 80% of lung cancer, which is a kind of cancer with high heterogeneity, so the genetic heterogeneity and molecular subtype should be explored.
Methods
Partitioning Around Medoid algorithm was used to acquire the molecular subtype for early-stage NSCLC based on prognosis-related mRNAs and methylation sites. Random forest (RF) and support vector machine (SVM) were used to build prediction models for subtypes.
Results
Six prognosis-related subtypes for early-stage NSCLC, including 4 subtypes for lung squamous cell carcinoma (LUSC) and 2 subtypes for lung adenocarcinoma (LUAD), were identified. There were highly expressed and hypermethylated gene regions for LUSC-C1 and LUAD-C2, highly expressed region for LUAD-C1, and hypermethylated regions for LUSC-C3 and LUSC-C4. Molecular subtypes for LUSC were mainly determined by DNA methylation (14 mRNAs and 362 methylation sites). Molecular subtypes for LUAD were determined by both mRNA and DNA methylation information (143 mRNAs and 458 methylation sites). Ten methylation sites were selected as biomarkers for prediction of LUSC-C1 and LUSC-C3, respectively. Nine genes and 1 methylation site were selected as biomarkers for LUAD subtype prediction. These subtypes can be predicted by the selected biomarkers with RF and SVM models.
Conclusions
In conclusion, we proposed a prognosis-related molecular subtype for early-stage NSCLC, which can provide important information for personalized therapy of patients.
Similar content being viewed by others
Background
Non-small cell lung carcinoma (NSCLC) accounts for more than 80% of lung cancer, which is the second most common cancer and the most common cause of cancer-associated deaths worldwide [1, 2]. With the development of diagnostic techniques, more NSCLC patients will be diagnosed at earlier stage [3, 4]. These patients can achieve a relatively superior prognosis, but some patients still develop recurrent cancer and about 40% of them will die of cancer within 5 years [5, 6]. NSCLC is also a kind of cancer with high heterogeneity, of which 45% were lung squamous cell carcinoma (LUSC) and 30% were lung adenocarcinoma (LUAD) [7]. Histological and genetic diversity can account for some of the individual variation in NSCLC survival. Therefore, identification of molecular subtype for early-stage NSCLC patients associated with survival will benefit early treatment and patient prognosis.
Molecular subtype has been used in the exploration of NSCLC heterogeneity. Gene expression subtypes of LUSC and LUAD have been proposed by The Cancer Genome Atlas (TCGA) research network, respectively [8, 9]. A multiplatform-based NSCLC molecular subtype including 9 subtypes for 1023 NSCLC patients has also been identified in a recent study [10]. There are some other kinds of lung cancer molecular subtypes according to different gene sets [11, 12]. However, there were some special molecular characteristics for early-stage NSCLC. Patient prognostic information has also not well utilized in these subtypes and gene sets, leading to weak predictive ability for patient prognosis.
In this study, we analyzed gene expression and DNA methylation data for early-stage NSCLC, and proposed a prognosis-related molecular subtypes for LUSC and LUAD. Then, we explored the function of differentially expressed genes and differentially methylated genes. We also selected biomarkers and built prediction model for each subtype in training dataset, and validated the models in test dataset. The prediction model was evaluated by sensitivity (SE), specificity (SP) and area under the ROC curve (AUC). Furthermore, we analyzed the molecular functions of these biomarkers in cancer development.
Methods
Datasets and preprocessing
RNA-Seq data, DNA methylation data and clinical information of NSCLC patients were downloaded from the UCSC Xena website (http://xena.ucsc.edu/). The RNA-seq data were log2 transformed RSEM normalized counts and mapped to HUGO gene symbols. The DNA methylation levels were represented by β-values (from 0 to 1). Methylation sites were filtered by the following criteria: 1) probes located in the X or Y chromosome; 2) SNP present within the assay of probe; 3) probes did not annotate with any reference genes; 4) probes located in the shelves and oversea regions of CpG island. Genes and methylation sites with missing value in more than 20% of patients were excluded, and patients without mRNA data or methylation data were also removed from further analysis. Data were centralized and standardized before analysis.
Molecular subtypes related with overall survival
For each gene and methylation site in the entire data set, we built a univariate Cox proportional hazard (Cox-PH) model and selected variables with P values less than 0.001. We than used these genes and methylation sites to cluster the patients using Partitioning Around Medoid (PAM) clustering algorithm. The cluster number K of PAM clustering algorithm was set to 2–5. The optimal number of NSCLC clusters was determined by maximizing the difference of overall survival among different subtypes. The Database for Annotation, Visualization and Integrated Discovery (version 6.8, DAVID) tool was used for the functional annotation for Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Prediction model for molecular subtypes
We randomly divided the data set into training set and test set, in which training set contained 60% patients (Table 1). We selected biomarkers and built prediction models for molecular subtypes in training set, and validated the models in test set. In the biomarker selection phase, univariate Wilcoxon test was firstly used to selected differentially expressed genes and methylated sites (P < 0.001) compared with other subtypes in the training dataset. Then, a multivariate partial least square (PLS) model was established, and 10 variables with largest variable important projection (VIP) values were selected as biomarkers for each subtype. Random forest (RF) and support vector machine (SVM) models were constructed with 10 selected biomarkers in training dataset. The model prediction ability was evaluated in training and test datasets, respectively.
Evaluation criteria
The prediction model performance was evaluated by sensitivity (SE), specificity (SP) and area under the ROC curve (AUC). SE and SP were defined by:
where.
True Positive (TP): the patient belongs to a subtype, and the prediction model predicts the patient as this subtype;
False Positive (FP): the patient does not belong to a subtype, but the prediction model predicts the patient as this subtype;
True Negative (TN): the patient does not belong to a subtype, and the prediction model does not predict the patient as this subtype;
False Negative (FN): the patient belongs to a subtype, but the prediction model does not predict the patient as this subtype.
AUC were defined by:
where n0 and n1 are the number of patients who belong to and not belong to a subtype respectively, and ri is the rank of ith patient of a subtype in the ranked list.
Results
Prognosis-related molecular subtypes
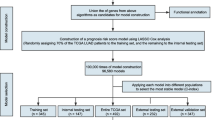
An overview workflow of this study was shown in Fig. 1. In total, 303 LUSC and 351 LUAD patients with coupled mRNA and methylation data were analyzed in this study. After screening according to the criteria in the “method” section, 16249 mRNAs and 162,926 methylation sites were included. In the univariate Cox regression analysis, 14 mRNAs and 362 methylation sites in LUSC, and 143 mRNAs and 458 methylation sites in LUAD were associated with overall survival at the 0.001 level.

An overview workflow of prognosis-related molecular subtypes identification and biomarkers selection in early-stage NSCLC
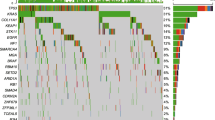
In the identification of prognosis-related molecular subtypes, the genes and methylation sites related with overall survival were used in the PAM clustering analysis, with cluster number K ranging from 2 to 5 (Supplementary Fig. 1). In the Kaplan-Meier (K-M) survival curves of LUSC and LUAD patients for different cluster number K, optimum survival curves were identified by 4 clusters for LUSC and 2 clusters for LUAD, respectively (Fig. 2). These subtypes showed different gene expression and DNA methylation patterns (Fig. 3). There were highly expressed and hypermethylated gene regions for LUSC-C1 and LUAD-C2, highly expressed region for LUAD-C1, and hypermethylated regions for LUSC-C3 and LUSC-C4.

The association between molecular subtypes and overall survival. a LUSC. b LUAD

The heatmap plot of mRNAs and methylation sites in different subtypes of NSCLC. Highly expressed and hypermethylated gene regions were marked with green boxes
Functional annotation of differentially expressed genes and methylated genes related with overall survival
Functional enrichment was conducted for genes and methylated genes related with overall survival in LUSC and LUAD. Firstly, 362 and 458 methylation sites in LUSC and LUAD were located in 267 and 339 genes, respectively. The distribution of methylation sites was showed in Supplementary Fig. 2. The methylated genes in LUSC were significantly enriched in 8 GO biological process terms, 4 GO cellular component terms, and 7 GO molecular function terms (Supplementary Table 1). The 14 differentially expressed genes were also enriched in 3 GO cellular component terms at 0.2 level.
For LUAD patients, the methylated genes were significantly enriched in 11 biological process terms, 9 cellular component terms, 7 molecular function terms, and 2 KEGG pathways (Supplementary Table 2). The differentially expressed genes were significantly enriched in 21 biological process terms, 12 cellular component terms, 11 molecular function terms, and 7 KEGG pathways.
Biomarkers for the prediction of subtypes
We next sought to select specific biomarkers and build prediction models for molecular subtypes. There were only 4 patients and obvious hypermethylated region in LUSC-C4 subtype, so we selected biomarkers and built prediction model for LUSC-C1, LUSC-C3 and LUAD. In the training dataset which included 60% patients, univariate Wilcoxon test was firstly used, and 114, 182, 303 biomarkers with P < 0.001 were selected for LUSC-C1, LUSC-C3 and LUAD. Then, these biomarkers were further used to build a multivariate PLS model and 10 variables with VIP values were selected as biomarkers for each subtype, respectively (Table 2). Ten methylation sites were selected as biomarkers for prediction of LUSC-C1 and LUSC-C3, respectively. Seven biomarkers (cg00894870, cg03041700, cg04738309, cg08356572, cg09844983, cg11416447 and cg22627950) were down-regulated, and 3 biomarkers (cg15987088, cg22627950 and cg24599434) were up-regulated in LUSC-C1 (Supplementary Fig. 3). Two biomarkers (cg02074191 and cg12065562) were down-regulated, and 8 biomarkers (cg00431236, cg00894870, cg02590972, cg03041700, cg04417954, cg08356572, cg11416447 and cg22627950) were up-regulated in LUSC-C3 (Supplementary Fig. 4). Nine genes and 1 methylation site were selected as biomarkers for LUAD subtype prediction. These biomarkers were all down-regulated in LUAD-C1 (Supplementary Fig. 5).
To evaluate the predictive performance of biomarkers, RF and SVM models for subtype prediction were built, and SE, SP and AUC were calculated in training and test datasets (Table 3). As results, we got a good prediction performance in the both training and test datasets. AUCs for LUSC-C1, LUSC-C3 and LUAD in RF model were 0.84, 0.77 and 0.83, whereas 0.85, 0.85 and 0.83 in SVM model, respectively. The prediction model built by SVM model was superior to the one built by RF model. These subtypes were significantly associated with overall survival, except for LUAD subtypes in test set, which also tended to be significant (Supplementary Fig. 6).
Discussion
In this study, we proposed a prognosis-related molecular subtype for early-stage NSCLC, including 4 subtypes for LUSC and 2 subtypes for LUAD. These subtypes showed different trend in overall survival, gene expression pattern, and DNA methylation level. Most subtypes showed highly expressed and hypermethylated gene regions, which facilitated the biomarker selection for subtypes. We also selected biomarkers and built prediction models with good performance, which can help the grouping of new patients and therapy strategy selection.
LUSC patients were divided into 4 clusters by 14 mRNAs and 362 methylation sites related with overall survival. These subtypes were mainly determined by DNA methylation information, and all the selected biomarkers were also methylation sites. Five methylation sites (cg00894870, cg03041700, cg08356572, cg11416447 and cg22627950) were selected as biomarkers for both LUSC-C1 and LUSC-C3, in which the function of 4 methylated genes were associated with cancer [13,14,15,16,17,18]. The function of 267 genes were mainly associated with regulation of cell cycle and gene transcription.
In LUSC-C1, 3 hyper-methylated sites were located in transcriptional start site (TSS) 200 regions of GHSR and weakly negatively related with GHSR (Supplementary Table 3), which can encode growth hormone secretagogue receptor (GHS-R) and related with energy metabolism. KIAA0090, ATAD3B, TRIM27 and DMTF1, regulated by hypo-methylated sites, were also associated with cancer. KIAA0090, which was positively related with hypo-methylated cg00894870, was associated with cancer metastasis and prognosis [16]. ATAD3B was expressed in cancer cell, and may related with tumorigenesis, proliferation and chemoresistance [14, 15]. TRIM27 was an oncogene [18] and DMTF1 can regulated ARF-p53 pathway [13, 17].
In LUSC-C3, 8 hyper-methylated sites were located in 10 genes. In addition to 4 same genes (KIAA0090, ATAD3B, TRIM27 and DMTF1) with LUSC-C1, ACP1 and SH3YL1 also played important roles in cancer. ACP1 can encode a tyrosine phosphatase, which was an anti-tumorigenic factor interacted with PDGF-R and FAK [19]. SH3YL1 can regulate migration of cancer cell [20]. Two hypo-methylated sites were located in gene body of PCDH gene family (PCDHA, PCDHB and PCDHG). The aberrant methylations of these genes were also found in breast cancer [21].
Unlike LUSC, LUAD patients were divided into 2 clusters by 143 mRNAs and 458 methylation sites, which indicated that these subtypes were determined by both mRNA and DNA methylation. These differentially expressed genes were mainly associated with cell cycle regulation. Whereas the differentially methylated genes were involved in a variety of GO terms and KEGG pathways, such as signal transduction, cell division and apoptosis.
In LUAD-C1, 10 selected biomarkers were all down-regulated in LUAD-C1. ANLN, CCNA2, CDCA5, DLGAP5, TPX2 and KIF4A were involved in the regulation of cell cycle (Supplementary Table 4). CKAP2L and SHCBP1 were associated with spindle formation, which was also involved in cell cycle. In previous study, over expression of 9 selected gene biomarkers (ANLN, CCNA2, CDCA5, CKAP2L, DLGAP5, KIF4A, KPNA2, SHCBP1 and TPX2) can indicate poor prognosis in different cancer types, including lung cancer, colon cancer, breast cancer and bladder cancer [22,23,24,25,26,27,28,29,30,31].
We built 2 prediction models for subtype prediction based on RF and SVM algorithms. The SE, SP and AUC for subtype prediction in training dataset were 1 by RF model, larger than the values calculated by SVM model. However, these values were smaller than those calculated by SVM model in test dataset. This phenomenon indicated that the model built by RF was over-fitting, and the prediction ability was worse for new data than SVM model.
Conclusions
In conclusion, we identified 6 subtypes for early-stage NSCLC, including 4 subtypes for LUSC and 2 subtypes for LUAD, by gene expression and DNA methylation data integration analysis. Furthermore, we also selected biomarkers and built prediction model to distinguish these subtypes, and most of these biomarkers were involved in tumor related function.
Availability of data and materials
Public access to the databases of this study is open. The data that support the findings of this study are available at https://xenabrowser.net/datapages/?cohort=TCGA%20Lung%20Adenocarcinoma%20(LUAD) and https://xenabrowser.net/datapages/?cohort=TCGA%20Lung%20Squamous%20Cell%20Carcinoma%20(LUSC).
Abbreviations
- AUC:
-
Area under the ROC curve
- DAVID:
-
The Database for Annotation, Visualization and Integrated Discovery
- FN:
-
False negative
- FP:
-
False positive
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- NSCLC:
-
Non-small cell lung carcinoma
- PLS:
-
Partial least square
- RF:
-
Random forest
- SE:
-
Sensitivity
- SP:
-
Specificity
- SVM:
-
Support vector machine
- TCGA:
-
The Cancer Genome Atlas
- TN:
-
True negative
- TP:
-
True positive
- VIP:
-
Variable important projection
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Liang W, Zhao Y, Huang W, Gao Y, Xu W, Tao J, Yang M, Li L, Ping W, Shen H, et al. Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics. 2019;9(7):2056–70.
Yang W, Qian F, Teng J, Wang H, Manegold C, Pilz LR, Voigt W, Zhang Y, Ye J, Chen Q, et al. Community-based lung cancer screening with low-dose CT in China: results of the baseline screening. Lung Cancer. 2018;117:20–6.
Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ, Wu YL, Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311.
Detterbeck FC, Chansky K, Groome P, Bolejack V, Crowley J, Shemanski L, Kennedy C, Krasnik M, Peake M, Rami-Porta R. The IASLC lung Cancer staging project: methodology and validation used in the development of proposals for revision of the stage classification of NSCLC in the forthcoming (eighth) edition of the TNM classification of lung Cancer. J Thorac Oncol. 2016;11(9):1433–46.
Duma N, Santana-Davila R, Molina JR. Non-small cell lung Cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. 2019;94(8):1623–40.
The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25.
The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50.
Chen F, Zhang Y, Parra E, Rodriguez J, Behrens C, Akbani R, Lu Y, Kurie JM, Gibbons DL, Mills GB, et al. Multiplatform-based molecular subtypes of non-small-cell lung cancer. Oncogene. 2016;36(10):1384–93.
Sherafatian M, Arjmand F. Decision tree-based classifiers for lung cancer diagnosis and subtyping using TCGA miRNA expression data. Oncol Lett. 2019;18(2):2125–31.
Dama E, Melocchi V, Dezi F, Pirroni S, Carletti RM, Brambilla D, Bertalot G, Casiraghi M, Maisonneuve P, Barberis M, et al. An aggressive subtype of stage I lung adenocarcinoma with molecular and prognostic characteristics typical of advanced lung cancers. Clin Cancer Res. 2017;23(1):62–72.
Ali M, Takayuki S, Pankaj T, Matise LA, Frazier DP, Mayur C, Hawkins GA, D'Agostino RB, Willingham MC, Kazushi I. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell. 2007;12(4):381–94.
Li S, Rousseau D. ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J Bioenerg Biomembr. 2012;44(1):189–97.
Merle N, Féraud O, Gilquin B, Hubstenberger A, Kieffer-Jacquinot S, Assard N, Bennaceur-Griscelli A, Honnorat J, Baudier J. ATAD3B is a human embryonic stem cell specific mitochondrial protein, re-expressed in cancer cells, that functions as dominant negative for the ubiquitous ATAD3A. Mitochondrion. 2012;12(4):441–8.
Molina RAS, Silva RR, Souza JF, Lopes MM, Tiezzi DG, Espreafico EM. Abstract C6: EMC1/KIAA0090 overexpression enhances proliferation and motility in breast cancer cells. Cancer Res. 2013;73(3 Supplement):C6.
Inoue K, Fry EA. Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int J Cancer. 2016;139(1):33–41.
Zhang Y, Feng Y, Ji D, Wang Q, Fu Z. TRIM27 functions as an oncogene by activating epithelial-mesenchymal transition and p-AKT in colorectal cancer. Int J Oncol. 2018;53(2):620–32.
Qiu X, Hu B, Huang Y, Deng Y, Wang X, Zheng F. Hypermethylation of ACP1, BMP4, and TSPYL5 in hepatocellular carcinoma and their potential clinical significance. Dig Dis Sci. 2016;61(1):149–57.
Kobayashi M, Harada K, Negishi M, Katoh H. Dock4 forms a complex with SH3YL1 and regulates cancer cell migration. Cell Signal. 2014;26(5):1082–8.
Petr N, Taylor J, Oshiro MM, Watts GS, Kim CJ, Futscher BW. Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res. 2008;68(20):8616.
Chie S, Yataro D, Nobuhisa I, Tatsuya K, Satoshi H, Tomoo I, Eiju T, Yusuke N. ANLN plays a critical role in human lung carcinogenesis through the activation of RHOA and by involvement in the phosphoinositide 3-kinase/AKT pathway. Cancer Res. 2005;65(24):11314.
Masaya T, Atsushi T, Nobuhisa I, Wataru Y, Kouki I, Hitoshi N, Eiju T, Nobuoki K, Yusuke N, Yataro D. Activation of KIF4A as a prognostic biomarker and therapeutic target for lung cancer. Clin Cancer Res. 2007;13(22 Pt 1):6624.
Wei P, Zhang N, Xu Y, Li X, Shi D, Wang Y, Li D, Cai S. TPX2 is a novel prognostic marker for the growth and metastasis of colon cancer. J Transl Med. 2013;11(1):313.
Christiansen A, Dyrskjøt L. The functional role of the novel biomarker karyopherin α 2 (KPNA2) in cancer. Cancer Lett. 2013;331(1):18–23.
Tian G, Yong H, Ling Y, Sheng A, Ziyu L, Jiafu J. CCNA2 is a prognostic biomarker for ER+ breast cancer and tamoxifen resistance. PLoS One. 2014;9(3):e91771.
Han-Seong K, Jae-Soo K, Yong-Bock C, Jungsil R, Hyun-Kyoung K, Mi-Kyung K, Byung-Ho N, Kyung-Tae K, Vishal C, Hye-Sil S. Chromatin CKAP2, a new proliferation marker, as independent prognostic indicator in breast cancer. PLoS One. 2014;9(6):e98160.
Chang IW, Lin VC, He HL, Hsu CT, Li CC, Wu WJ, Huang CN, Wu TF, Li CF. CDCA5 overexpression is an indicator of poor prognosis in patients with urothelial carcinomas of the upper urinary tract and urinary bladder. Am J Transl Res. 2015;7(4):710–22.
Feng W, Li HC, Xu K, Chen YF, Pan LY, Mei Y, Cai H, Jiang YM, Chen T, Feng DX. SHCBP1 is over-expressed in breast cancer and is important in the proliferation and apoptosis of the human malignant breast cancer cell line. Gene. 2016;587(1):91–7.
Schneider MA, Christopoulos P, Muley T, Warth A, Klingmueller U, Thomas M, Herth FJF, Dienemann H, Mueller NS, Theis F. AURKA, DLGAP5, TPX2, KIF11 and CKAP5: five specific mitosis-associated genes correlate with poor prognosis for non-small cell lung cancer patients. Int J Oncol. 2017;50(2):365–72.
Phan NN, Wang CY, Li KL, Chen CF, Chiao CC, Yu HG, Huang PL, Lin YC. Distinct expression of CDCA3, CDCA5, and CDCA8 leads to shorter relapse free survival in breast cancer patient. Oncotarget. 2018;9(6):6977–92.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81703322) and China Postdoctoral Science Foundation (No. 2020 M673082) financed study designing, data collection, data analysis and writing the manuscript.
Author information
Authors and Affiliations
Contributions
YW designed the study. KY and YW analyzed the data and wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The data used in this study were all from the publicly accessible TCGA database. Ethics committee approval was not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Fig. 1
. Survival curves for different number of molecular subtypes. Supplementary Fig. 2. The distribution of methylation sites. Supplementary Fig. 3. Boxplots of 10 methylation site biomarkers for LUSC-C1. Supplementary Fig. 4. Boxplots of 10 methylation site biomarkers for LUSC-C3. Supplementary Fig. 5. Boxplots of 9 gene expression and 1 methylation site biomarkers for LUAD-C1. Supplementary Fig. 6.The K-M survival curve for molecular subtypes and overall survival in different data set. (A) LUSC in training set. (B) LUAD in training set. (C) LUSC in test set. (D) LUAD in test set. Supplementary Table 1. Functional enrichment of differentially expressed genes and methylated genes related with overall survival for LUSC. Supplementary Table 2. Functional enrichment of differentially expressed genes and methylated genes related with overall survival for LUAD. Supplementary Table 3. The correlation of methylation sites and located genes. Supplementary Table 4. Functional enrichment of 9 biomarkers for LUAD-C1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, K., Wu, Y. A prognosis-related molecular subtype for early-stage non-small lung cell carcinoma by multi-omics integration analysis. BMC Cancer 21, 128 (2021). https://doi.org/10.1186/s12885-021-07846-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-07846-0