
Abstract
Background
Impaired glucose metabolism–related genetic variants and traits likely interact with obesity and related lifestyle factors, influencing postmenopausal breast and colorectal cancer (CRC), but their interconnected pathways are not fully understood. By stratifying via obesity and lifestyles, we partitioned the total effect of glucose metabolism genetic variants on cancer risk into two putative mechanisms: 1) indirect (risk-associated glucose metabolism genetic variants mediated by glucose metabolism traits) and 2) direct (risk-associated glucose metabolism genetic variants through pathways other than glucose metabolism traits) effects.
Method
Using 16 single-nucleotide polymorphisms (SNPs) associated with glucose metabolism and data from 5379 postmenopausal women in the Women’s Health Initiative Harmonized and Imputed Genome-Wide Association Studies, we retrospectively assessed the indirect and direct effects of glucose metabolism-traits (fasting glucose, insulin, and homeostatic model assessment–insulin resistance [HOMA-IR]) using two quantitative tests.
Results
Several SNPs were associated with breast cancer and CRC risk, and these SNP–cancer associations differed between non-obese and obese women. In both strata, the direct effect of cancer risk associated with the SNP accounted for the majority of the total effect for most SNPs, with roughly 10% of cancer risk due to the SNP that was from an indirect effect mediated by glucose metabolism traits. No apparent differences in the indirect (glucose metabolism-mediated) effects were seen between non-obese and obese women. It is notable that among obese women, 50% of cancer risk was mediated via glucose metabolism trait, owing to two SNPs: in breast cancer, in relation to GCKR through glucose, and in CRC, in relation to DGKB/TMEM195 through HOMA-IR.
Conclusions
Our findings suggest that glucose metabolism genetic variants interact with obesity, resulting in altered cancer risk through pathways other than those mediated by glucose metabolism traits.
Similar content being viewed by others


Background
Breast cancer is the most commonly occurring cancer and the second most common cause of cancer-related deaths in the United States [1]. Colorectal cancer (CRC) is the second most commonly diagnosed cancer and one of the leading causes of cancer-related mortality throughout the world [2]. Impaired glucose metabolism, i.e. insulin resistance (IR), is characterized by hyperinsulinemia and hyperglycemia, and demonstrates strong associations with breast cancer and CRC [3,4,5,6,7,8]. The association is particularly strong in postmenopausal women, in whom high insulin levels have been associated with a twofold increase in breast cancer risk [9, 10]. The homeostatic model assessment–insulin resistance (HOMA-IR) reflecting high blood levels of insulin and glucose is positively associated with breast cancer in the postmenopausal women [11].
Besides its importance in glucose homeostasis, insulin is an essential hormone in anabolic processes in early cell growth and development, directly through the insulin receptor and indirectly through the insulin-like growth factor receptor [12, 13]. Insulin receptors that are mainly found in adipose tissues, muscle, and liver cells are overexpressed in breast cancer and CRC cells. This overexpression results in the enhanced anabolic state necessary for cell proliferation, differentiation, and anti-apoptosis, via abnormal stimulation of multiple signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)/serine/threonine-specific protein kinase (Akt) and mitogen-activated protein kinase (MAPK) pathways [14, 15]. In addition, high glucose levels owing to glucose intolerance induce high levels of intracellular glucose, facilitating breast cancer and CRC cell growth [6, 8]. Thus, impaired glucose metabolism, such as IR, leading to hyperglycemia and hyperinsulinemia, contributes to overexpression of these receptors and multiple abnormal cellular signaling cascades, and therefore may be associated with carcinogenesis. Considering the relationships of these glycemic phenotypes and cancer risk, the glucose metabolism-related genetic variants that are related to impaired glucose metabolic syndromes (e.g. high glucose, insulin, and HOMA-IR levels) are plausibly associated with increased risk of breast cancer and CRC. A limited number of population-based epidemiologic studies have been performed to examine these relationships [16,17,18,19,20,21,22].
Breast cancer, particularly in postmenopausal women, and CRC risk are elevated among those who are obese [4, 23,24,25,26]. Obesity status and obesity-related lifestyle factors are accompanied by elevated glucose metabolism traits (e.g., insulin, glucose, and HOMA-IR levels) [4, 23, 24]. Specifically, physical inactivity and high-fat diet, as modifiable factors for obesity, [3] increase insulin levels and IR, and are associated with increased risk of breast cancer [8, 27, 28] and CRC [29,30,31,32]. Further, previous in vitro studies have revealed obesity– glucose metabolism-related gene signature–breast cancer or CRC risk pathways, suggesting that glucose metabolism-related genetic variants interact with obesity and jointly influence cancer susceptibility [15, 27, 33,34,35,36].
In this study among postmenopausal women, we examined the pathway of glucose metabolism genetic variants, glucose metabolism traits (fasting insulin, glucose, and HOMA-IR levels), and cancer risk. We focused on the mediation effects relating glucose metabolism genetic variants (exposure) and breast cancer and CRC risk (outcome), and on the role of glucose metabolism traits (mediator) that play in this association (Fig. 1). We first evaluated the magnitude of the total effect of glucose metabolism genetic variants on breast cancer and CRC (i.e. the overall genetic effect, without considering the effect of glucose metabolism traits). We then evaluated how this total effect is partitioned into direct (cancer risk associated with glucose metabolism genetic variants through pathways other than glucose metabolism traits) and indirect effects (cancer risk associated with glucose metabolism genetic variants through pathways mediated by glucose metabolism traits). This approach allowed us to test the hypothesis that glucose metabolism-related genetic variants are associated with increased risk of cancers and that the relationships depend on impaired glucose metabolism symptoms (high insulin, glucose, and HOMA-IR levels).

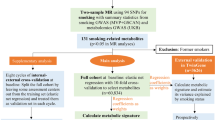
Diagrams of total, direct, and indirect pathways of SNPs in glucose metabolism genes, glucose metabolism traits, and cancer risk. (HOMA-IR, homeostatic model assessment–insulin resistance; HR, hazard ratio; SNP, single-nucleotide polymorphism.). a C is a total effect (overall genetic effect, without considering the effect of glucose metabolism traits), expressed via HR. b C′ is a direct effect (cancer risk associated with glucose metabolism-relevant genetic variants through pathways other than glucose metabolism traits), expressed via HR after accounting for mediator; a*b (≈C-C′) is an indirect effect (cancer risk associated with glucose metabolism-relevant genetic variants through pathways mediated by glucose metabolism traits)
Given that the association between glucose-metabolism genetic factors and glucose-metabolism traits could be influenced by obesity [4, 8, 23, 24, 27,28,29,30,31,32], and through this glycemic mechanism, obesity status and related factors are associated with breast cancer and CRC [15, 27, 33,34,35,36], we evaluated how the pathway of glucose metabolism genetic factors, glucose metabolism traits, and cancer is influenced by obesity and obesity-related factors. We examined whether glucose metabolism genetic variants’ interactions with obesity and relevant lifestyle factors influence glucose metabolism traits and whether these changes in traits alter the association between glucose metabolism traits and cancer risk. Further, we assessed whether these altered relationships (glucose metabolism gene–glucose metabolism traits relationship and glucose metabolism traits–cancer risk relationship) influence the association between glucose metabolism genetic variants and cancer risk.
Disentangling these complicated gene–phenotype–lifestyle interactions will provide insights into the role of glucose intolerance in the development of obesity-related breast cancer and CRC and suggest strategies to reduce cancer risk in postmenopausal women.
Methods
Study population
This study included data from 5379 participants enrolled in the Women’s Health Initiative (WHI) Harmonized and Imputed Genome-Wide Association Studies (GWAS), which is the effort of a joint imputation and harmonization effort for GWAS within the WHI Clinical Trials and Observational Studies. Details of this study’s rationale and design have been described elsewhere [37, 38]. Briefly, WHI study participants were recruited from 40 clinical centers nationwide between October 1, 1993, and December 31, 1998. Eligible women were 50–79 years old, postmenopausal, expected to live near the clinical centers for at least 3 years after enrollment, and able to provide written consent. For our study, we included only European-American women. From among the 7835 women who did not have diabetes mellitus (DM) at enrollment or later, and had at least 8 hours’ fasting glucose and/or insulin concentrations available at baseline, we excluded women who had been followed up for less than 1 year or those diagnosed with any cancer at enrollment, resulting in 6748 participants. We excluded another 1369 women whose information on covariates was unavailable, leaving a final total of 5379 women (80% of the eligible 6748). This study was approved by the institutional review boards at the University of California, Los Angeles.
Data collection and outcome variables
Standardized written protocols had been used and periodic quality assurance performed by the WHI coordinating center to ensure uniform data collection. At baseline, participants had completed self-questionnaires on demographic and lifestyle factors and their medical and reproductive histories. Anthropometric measurements, including height, weight, and waist and hip circumferences had been obtained at baseline by trained staff. Of 33 variables initially chosen from a literature review for their associations with glucose metabolism and breast cancer and CRC, we selected 29 final variables (Table 1) for this study after performing univariate and stepwise regression analyses and multicollinearity testing.
Cancer outcomes were determined via a centralized review of medical charts, and cancer cases were coded according to the National Cancer Institute’s Surveillance, Epidemiology, and End-Results guidelines [39]. The outcome variables were the specific cancer type (breast cancer and CRC) and the time to develop such cancer. The time from enrollment to cancer development, censoring, or study end-point was recorded as the number of days and then converted into years.
Genotyping and laboratory methods
The WHI imputed GWAS comprises six substudies (Hip Fracture GWAS, SHARe, GARNET, WHIMS, GECCO, and MOPMAP) within the WHI study. Participants provided DNA samples at baseline and genotyping included alignment (“flipping”) to the same reference panel and imputation via the 1000 Genomes reference panels. Single-nucleotide polymorphisms (SNPs) for harmonization were checked for pairwise concordance among all samples in the substudies. Initial quality assurance was conducted according to a standardized protocol, with a missing call rate of <2% and Hardy-Weinberg Equilibrium of p ≥ 10−4. Sixteen SNP candidates, available for this study with 97% R-squared imputation quality scores, were selected on the basis of their association (p < 5 × 10−8) with fasting glucose and/or insulin concentrations in a previous meta-analysis with independent replication [40,41,42].
Fasting blood samples had been collected from each participant at baseline by trained phlebotomists and immediately centrifuged and stored at −70 °C. Serum glucose was measured using the hexokinase method on a Hitachi 747 analyzer (Boehringer Mannheim Diagnostics), with coefficient of variation of 1.6% and correlation coefficient of values of 0.99. Serum insulin testing had been conducted by Sandwich Immunoassay on a Roche Elecsys 2010 analyzer (Roche Diagnostics). The coefficient of variation and correlation coefficient of values for insulin were 4.9% and 0.99, respectively. HOMA-IR was estimated as glucose (unit: mg/dl) × insulin (unit: μIU/ml) / 405 [43].
Statistical analysis
Participants’ differences in baseline characteristics, stratified by obesity status (body mass index [BMI], waist circumference, and waist-to-hip ratio [w/h]), level of physical activity, and dietary fat intake, were assessed by using unpaired two-sample t tests for continuous variables, and chi-squared tests for categorical variables. If continuous variables were skewed or had outliers, Wilcoxon’s rank-sum test was implemented. With the regression assumptions met, multiple linear regression was performed to produce effect sizes and 95% confidence intervals (CIs) of the exposure (glucose metabolism-related SNPs with an additive and dominant model) to predict the outcomes (fasting glucose, insulin, and HOMA-IR levels) (Additional file 1: Tables S1.1–6).
The Cox proportional hazards regression model was used to obtain hazard ratios (HRs) and 95% CIs for glucose, insulin, and HOMA-IR levels and glucose metabolism-related SNPs in predicting breast cancer and CRC. The proportional hazards assumption was tested via a Schoenfeld residual plot and rho. The model was adjusted for covariates (e.g., age, education, family history of DM and cancer, comorbidity, lifestyle factors including smoking, physical activity, depression, lifetime partner, and diet, obesity, and reproductive history).
A direct and total effect size of glucose metabolism-related SNP (exposure) on breast cancer and CRC (outcome) was produced from the HR for glucose metabolism-related SNP on cancer in the Cox model that included all covariates, with (direct) and without (total) glucose, insulin, and HOMA-IR levels (mediator). The mediation effect size and testing for its significance (i.e. the pathway of glucose metabolism-SNPs and cancer risk through insulin, glucose, and HOMA-IR levels) were produced via the use of two complementary statistical methods [44,45,46]: 1) bootstrapping the sampling distribution for standard errors using Mplus software and 2) the percentage change in the HRs by comparing a model that includes all covariates with a model that includes all covariates and the mediator [44, 45]. These two approaches, differently from traditional Baron-Kenny steps, enabled us not only to prevent results from being affected by Type II errors but also to estimate the amount and test the significance of the mediation effect [44]. To evaluate the role of obesity and correlated lifestyle factors as an effect modifier on the pathway of glucose metabolism genetic factors, glucose metabolism traits, and cancer, we stratified participants by those potential effect modifiers, and within the strata, compared the proportions of the cancer risk contributed by glucose metabolism genetic variants through the glucose metabolism traits (indirect effect) and non-glucose metabolism pathways (direct effect). A two-tailed p-value <0.05 was considered statistically significant. The R statistical package (v 2.15.1) was used.
Results
Participants’ baseline characteristics between non-obese (BMI <30.0) and obese (BMI ≥30.0) women are presented in Table 1. Obese women were younger, less educated, and more likely to have a history of hypertension and a family history of DM than non-obese women. Also obese women were less likely to be current smokers, and to meet the physical activity and dietary guidelines, and they were more likely to have higher percentages of calories from dietary fat intake. Further, more obese women tended to have a history of hysterectomy or oophorectomy and earlier menarche, and they were less likely to use exogenous estrogen. They also had higher serum levels of fasting glucose, insulin, and HOMA-IR. We stratified participants by waist circumference, w/h, level of physical activity, and dietary fat intake, using a cutoff value relevant to glucose intolerance, [47] and compared their characteristics (Additional file 2: Tables S2.1–4). The participants had been followed up through August 29, 2014 (a median follow-up period of 16 years), resulting in 326 participants (5% of non-obese and 8% of obese women) diagnosed with breast cancer, and 364 participants (6% of non-obese and 8% of obese women) diagnosed with CRC.
Sixteen SNPs were selected from previous GWAS as being associated with glucose metabolism traits. The allele frequencies of these SNPs in our population were consistent with frequencies of those in a European population [48]. No significant differences in allele frequency between strata (obesity, physical activity, and high-fat diet) were observed (Additional file 3: Tables S3.1–5).
Breast cancer risk associated with glucose metabolism-related SNPs mediated through glucose metabolism traits, stratified by obesity status (BMI, waist, and w/h), level of physical activity, and dietary fat intake
We partitioned the total effect of glucose metabolism-related SNPs on breast cancer risk into indirect (via glucose metabolism traits) and direct (not via glucose metabolism traits) effects. Each of these analyses was mediated by fasting glucose (Table 2), HOMA-IR (Table 3), and insulin levels (Additional file 4: Table S4.1). For each mediator, the glucose metabolism-SNP–cancer association was evaluated, stratified by obesity status (BMI < 30 vs. ≥ 30; waist ≤88 cm vs. > 88 cm; and w/h ≤ 0.85 vs. > 0.85), level of physical activity (metabolic equivalent [MET] ≥ 10 vs. < 10), and dietary fat intake (< 40% vs. ≥ 40% calories from fat).
Of the 16 candidate SNPs, three had significant associations with breast cancer risk. The SNP–cancer risk effect was stronger in each SNP for a direct effect than an indirect effect regardless of the mediator. Carriers of the G6PC2 rs560887 T minor-allele were associated with increased breast cancer risk in obese women, stratified by BMI, waist, w/h, and dietary fat intake (Tables 2 and 3, and Additional file 4: Table S4.1). Roughly 15% of the breast cancer risk owing to this genetic variant was mediated via glucose metabolism traits in the obese group; no significant differences in mediation effect were found between the obese and non-obese women.
Carriers of the IGF1 rs35767 A minor-allele had associations similar to those found in the carriers of G6PC2 (Tables 2 and 3, and Additional file 4: Table S4.1). Compared with the carriers in the non-obese group (w/h ≤ 0.85), in whom no significant association with cancer was found, the carriers in the obese group (w/h > 0.85) had an association with increased breast cancer risk; further, in this obese group, about 10% of the breast cancer risk associated with this genetic variant was dependent on glucose metabolism traits. In addition, no differences were apparent in mediation effect between women with w/h ≤ 0.85 and those with w/h > 0.85. Carriers of the GCKR rs780094 C major-allele had an association with increased risk of breast cancer in women with w/h > 0.85 (Table 2); approximately 50% of cancer risk attributable to this variant was mediated via glucose levels in this obese group.
CRC risk associated with glucose metabolism-related SNPs mediated through glucose metabolism traits, stratified by obesity status (BMI, waist, and w/h), level of physical activity, and dietary fat intake
We also split the total effect of the CRC risk–glucose metabolism SNP relationship into direct and indirect effects through fasting glucose (Table 4), HOMA-IR (Table 5), and insulin levels (Additional file 4: Table S4.2). For each mediator, those effects were stratified by obesity status (BMI, waist, and w/h), level of physical activity, and dietary fat intake. Overall, the direct effect of glucose metabolism SNPs on increased CRC risk accounted for a majority of the total effect, suggesting a minimal influence of indirect effect on the total effect. In addition, the indirect effects mediated via glucose metabolism traits were not apparently different between obesity strata.
Carriers of the GCK rs4607517 G major-allele had an association with decreased CRC risk in non-obese women with BMI < 30 and MET ≥10, and in obese women with ≥40% calories from fat (see total effect in Tables 4 and 5). Compared with the total effects, the direct effects of glucose metabolism-related SNP on CRC risk, after accounting for glucose (Table 4) or HOMA-IR (Table 5), decreased slightly but were no longer statistically significant; it suggested existence of glucose metabolism traits’ mediation effects (roughly, 10%) on the SNP–cancer risk. Similarly, carriers of the CRY2 rs11605924 C major-allele had an association with decreased CRC risk in women with BMI < 30 and waist ≤88 cm (Tables 4 and 5); after accounting for glucose (Table 4) or HOMA-IR (Table 5), the direct effects were no longer significant, indicating potential mediation effects (roughly 5%) on the SNP–CRC risk association. In addition, carriers of the G6PC2 rs560887 T minor-allele had an association with decreased CRC risk in women with waist ≤88 cm, and the mediation effect of glucose on the SNP–CRC risk association in these non-obese carriers resulted in the decreased direct effect (roughly 15%) of CRC risk in relation to G6PC2 carriers (Table 4).
In contrast, carriers of the FADS1 rs174550 T major-allele, the ADRA2A rs10885122 G major-allele, and the SLC30A8 rs11558471 A major-allele had associations with increased CRC risk in obese women (BMI ≥ 30, waist >88 cm for FADS1 carriers; w/h > 0.85 for ADRA2A carriers; and ≥40% calories from fat for SLC30A8 carriers) (Tables 4 and 5, and Additional file 4: Table S4.2). Roughly, less than 10% of the CRC risk due to each genetic variant was mediated via glucose, HOMA-IR, or insulin in the relevant obese groups. No significantly different mediation effects were found between obesity strata. Likewise, carriers of the DGKB/TMEM195 rs2191349 G minor-allele had an association with increased risk of CRC in obese women (BMI ≥ 30, waist >88 cm, and w/h > 0.85) (Table 5 and Additional file 4: Table S4.2). The insulin effect as a mediator in these obese carriers was minimal (15%) (Additional file 4: Table S4.2). On the contrary, the HOMA-IR mediator effect in this group (Table 5) accounted for approximately 50% of the total effect. This resulted in the elevated and significant direct effect of SNP–CRC risk (i.e. from total effect after accounting for the mediators); it suggests a positive effect of HOMA-IR on the total effect of the SNP–CRC association.
Discussion
In this retrospective study of data from a large cohort of postmenopausal women, by using 16 glucose metabolism-related SNPs previously associated with glycemic metabolic traits, [40,41,42] we partitioned the total effect of glucose metabolism genetic variants on breast cancer and CRC into direct (cancer risk associated with SNPs mediated through pathways other than glucose metabolism traits) and indirect (cancer risk associated with SNPs mediated by glucose metabolism traits) effects. By stratifying data via obesity status and obesity-relevant lifestyle factors, we also assessed how those effects differed between strata. There have been relatively few population-based epidemiologic studies between glucose metabolism genetic variants and breast cancer and CRC risk [16,17,18,19,20,21,22]. To our knowledge, this is the first study to evaluate the association between glucose metabolism genetic variants and breast cancer and CRC risk by partitioning the glucose metabolism genetic variants’ effects on the risk for those cancers into direct and indirect effects. Additionally, we assessed the role of obesity and related factors as effect modifiers.
We found that among the16 glucose metabolism-related SNPs evaluated, three were associated with breast cancer risk, and seven with CRC risk. These SNPs’ associations with cancer risk differed between non-obese and obese carriers, indicating that glucose metabolism-related SNPs’ interactions with obesity and related lifestyle factors influence cancer risk. For most of the SNPs we studied, the direct effects on cancer risk accounted for a majority of the total effect: only roughly 15% of the cancer risk associated with glucose metabolism-related SNPs was mediated via glucose metabolism traits. This suggests that glucose metabolism traits are not the main mediators through which glucose metabolism-related SNPs are associated with increased risk for breast cancer and CRC. Further, no apparent differences in the indirect effects (mediated via glucose metabolism traits) were observed between non-obese and obese strata. Our findings thus indicate that glucose metabolism-related genetic variants interact with obesity and lifestyle factors, resulting in altered cancer risk not through glucose metabolism traits pathways, but through different mechanisms.
In relation to breast cancer risk, obese carriers of G6PC2, IGF1, and GCKR had an association with increased risk. Expression of the G6PC2 gene (glycolytic inhibitor) is elevated in cancer cells and related to a decreased survival rate in cancer patients, suggesting its role in glucose metabolism and cell cycle control in cancer cells [49,50,51]. The IGF1 and GCKR variants are related to glucose metabolism; both are highly expressed in the liver, contributing to hepatic glucose metabolism [41]. IGFI encodes insulin-like growth factor I, which is well known to increase cancer risk, and elevates HOMA-IR levels [22, 40]. Additionally, GCKR inhibits glucokinase, a key protein in glucose metabolism, leading to increased hepatic glucose production [41, 52]. These facts support the biological plausibility of the carriers’ association with increased breast cancer risk. Further, in this study, the carriers of these variants had association with breast cancer, but only among the obese women, suggesting that adiposity plays a strong role in modulating the effect of these variants on carcinogenesis. Interestingly, the mediation effects of glucose metabolism traits accounted for only a small portion of the overall the G6PC2– and IGF1–cancer associations in both non-obese and obese women, suggesting that different pathways exist through which obesity interacts with the G6PC2 and IGF1 genetic variants and breast cancer risk. In contrast, the GCKR variant’s effect on cancer was mediated through glucose by 50% in obese women (but not in non-obese women), indicating that an adiposity-related carcinogenetic pathway in this variant intermingles with the glucose-intolerance system.
Of the seven SNPs related to CRC risk, three (GCK, CRY2, and G6PC2) had a lower association with CRC among non-obese women. GCK opposing G6PC2 encodes for glucokinsase, and mutation of this gene is related to DM and glucose metabolism; further, the GCK variant is associated with prostatic and pancreatic cancers [53, 54]. Our study showed a reduced CRC risk in non-obese female carriers of this variant, indicating that a cancer-specific mechanism incorporating glucose metabolism traits and genes as well as obesity should be investigated. In addition, mutation of CRY2 results in dysfunction of circadian rhythms and is associated with tumorigenesis [20, 55]. Our finding of reduced CRC risk associated with the CRY2 variant in non-obese women warrants further study.
The other four of the seven CRC related SNPs in our study (FADS1, ADRA2A, SLC30A8, and DGKB/TMEM195) had an increased relationship with CRC among obese women. FADS1, which encodes fatty acid desaturase 1, produces arachidonic acid related to increased insulin. One earlier study [19] reported CRC risk associated with this genetic variant, and their results are consistent with ours. ADRA2A and SLC30A8 have not been studied for an association with CRC, but the functional changes that have been reported followed by mutations (in ADRA2A, modified insulin release by adrenergic suppression, and in SLC30A8, altered storage and maturation of insulin in beta cells [40, 56]) support our findings of increased CRC risk in relation to these variants. Finally, DGKB regulates diacylglycerol and potentiates insulin secretion, indicating that its mutation influences glucose homeostasis [40]; our findings suggest that this genetic variant is related to carcinogenesis in obese women.
Although obesity interacts with these seven SNPs and influences CRC risk differently between non-obese and obese carriers, the indirect effects of glucose metabolism traits on the SNP–CRC risk were minimal and did not differ between obesity strata (except in the case of DGKB/TMEM195). Further study is needed to examine obesity–glycemic gene–CRC mechanisms mediated through different pathways. In contrast, among obese women, roughly 50% of CRC risk associated with DGKB/TMEM195 variant was mediated via HOMA-IR. This supports the role of adiposity in carcinogenesis through deregulated glycemic metabolism.
We did not conduct any subtype analyses of breast cancer cases due to insufficient statistical power (cases represented less than 3% of each subset). Since we were using this analysis to generate new hypotheses, we did not include any multiple-testing adjustments in our analyses. On the basis of prior findings of 16 loci associated with glucose metabolism, we tested the hypothesis that these genetic variants’ interactions with obesity and lifestyle modifiers influence glucose homeostasis, resulting in altered cancer risk. The small indirect effect could be due to measurement error in the mediators. Since our study was conducted using data from only European-American postmenopausal women, care should be taken when generalizing our findings to other populations.
Conclusions
Our results suggest that in postmenopausal women, glucose intolerance has a potential role in the risk for breast cancer and CRC. Obesity modulates the glucose metabolism genetic variant–cancer risk association through pathways other than glucose metabolism traits. Further studies are needed to explore these complicated mechanisms. Our study provides insight into gene–lifestyle interactions and suggests data on potential genetic targets for use in clinical trials for cancer prevention and intervention strategies to reduce the cancer risk in postmenopausal women.
Abbreviations
- BMI:
-
Body mass index
- CI:
-
Confidence interval
- CRC:
-
Colorectal cancer
- DM:
-
Diabetes mellitus
- GWAS:
-
Genome-wide association studies
- HOMA-IR:
-
Homeostatic model assessment–insulin resistance
- HR:
-
Hazard ratio
- IR:
-
Insulin resistance
- MET:
-
Metabolic equivalent
- SNP:
-
Single-nucleotide polymorphism
- w/h:
-
Weight-to-hip ratio
- WHI:
-
Women’s health initiative
References
American Cancer Society. Breast Cancer Facts & Figures 2015–2016. Atlanta: American Cancer Society Inc.; 2015.
American Cancer Society. Global Cancer Facts & Figures 3rd Edition. Atlanta: American Cancer Society, Inc.;2015.
Tenesa A, Campbell H, Theodoratou E, Dunlop L, Cetnarskyj R, Farrington SM, Dunlop MG. Common genetic variants at the MC4R locus are associated with obesity, but not with dietary energy intake or colorectal cancer in the Scottish population. Int J Obes. 2009;33(2):284–8.
Pendyala S, Neff LM, Suarez-Farinas M, Holt PR. Diet-induced weight loss reduces colorectal inflammation: implications for colorectal carcinogenesis. Am J Clin Nutr. 2011;93(2):234–42.
Nimptsch K, Aleksandrova K, Boeing H, Janke J, Lee YA, Jenab M, Kong SY, Tsilidis KK, Weiderpass E, Bueno-De-Mesquita HB, et al. Plasma fetuin-a concentration, genetic variation in the AHSG gene and risk of colorectal cancer. Int J Cancer. 2015;137(4):911–20.
Lee SK, Moon JW, Lee YW, Lee JO, Kim SJ, Kim N, Kim J, Kim HS, Park SH. The effect of high glucose levels on the hypermethylation of protein phosphatase 1 regulatory subunit 3C (PPP1R3C) gene in colorectal cancer. J Genet. 2015;94(1):75–85.
Wairagu PM, Phan AN, Kim MK, Han J, Kim HW, Choi JW, Kim KW, Cha SK, Park KH, Jeong Y. Insulin priming effect on estradiol-induced breast cancer metabolism and growth. Cancer Biol Ther. 2015;16(3):484–92.
Wahdan-Alaswad R, Fan Z, Edgerton SM, Liu B, Deng XS, Arnadottir SS, Richer JK, Anderson SM, Thor AD. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle. 2013;12(24):3759–69.
Kabat GC, Kim M, Caan BJ, Chlebowski RT, Gunter MJ, Ho GY, Rodriguez BL, Shikany JM, Strickler HD, Vitolins MZ, et al. Repeated measures of serum glucose and insulin in relation to postmenopausal breast cancer. Int J Cancer. 2009;125(11):2704–10.
Vona-Davis L, Rose DP. Type 2 diabetes and obesity metabolic interactions: common factors for breast cancer risk and novel approaches to prevention and therapy. Curr Diabetes Rev. 2012;8(2):116–30.
Sieri S, Muti P, Claudia A, Berrino F, Pala V, Grioni S, Abagnato CA, Blandino G, Contiero P, Schunemann HJ, et al. Prospective study on the role of glucose metabolism in breast cancer occurrence. Int J Cancer. 2012;130(4):921–9.
Clayton PE, Banerjee I, Murray PG, Renehan AG. Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nat Rev Endocrinol. 2011;7(1):11–24.
Boyd DB. Insulin and cancer. Integr Cancer Ther. 2003;2(4):315–29.
Argiles JM, Lopez-Soriano FJ. Insulin and cancer (review). Int J Oncol. 2001;18(4):683–7.
Arcidiacono B, Iiritano S, Nocera A, Possidente K, Nevolo MT, Ventura V, Foti D, Chiefari E, Brunetti A. Insulin resistance and cancer risk: an overview of the pathogenetic mechanisms. Exp Diabetes Res. 2012;2012:789174.
Ollberding NJ, Cheng I, Wilkens LR, Henderson BE, Pollak MN, Kolonel LN, Le Marchand L. Genetic variants, prediagnostic circulating levels of insulin-like growth factors, insulin, and glucose and the risk of colorectal cancer: the multiethnic cohort study. Cancer Epidemiol Biomarkers Prev. 2012;21(5):810–20.
Feik E, Baierl A, Hieger B, Fuhrlinger G, Pentz A, Stattner S, Weiss W, Pulgram T, Leeb G, Mach K, et al. Association of IGF1 and IGFBP3 polymorphisms with colorectal polyps and colorectal cancer risk. Cancer Causes Control. 2010;21(1):91–7.
Kaabi B, Belaaloui G, Benbrahim W, Hamizi K, Sadelaoud M, Toumi W, Bounecer H. ADRA2A Germline Gene polymorphism is associated to the severity, but not to the risk, of breast cancer. Pathol Oncol Res. 2016;22(2):357–65.
Zhang B, Jia WH, Matsuda K, Kweon SS, Matsuo K, Xiang YB, Shin A, Jee SH, Kim DH, Cai Q, et al. Large-scale genetic study in east Asians identifies six new loci associated with colorectal cancer risk. Nat Genet. 2014;46(6):533–42.
Mazzoccoli G, Colangelo T, Panza A, Rubino R, De Cata A, Tiberio C, Valvano MR, Pazienza V, Merla G, Augello B, et al. Deregulated expression of cryptochrome genes in human colorectal cancer. Mol Cancer. 2016;15(1):6.
Mao Y, Fu A, Hoffman AE, Jacobs DI, Jin M, Chen K, Zhu Y. The circadian gene CRY2 is associated with breast cancer aggressiveness possibly via epigenomic modifications. Tumour Biol. 2015;36(5):3533–9.
Pechlivanis S, Wagner K, Chang-Claude J, Hoffmeister M, Brenner H, Forsti A. Polymorphisms in the insulin like growth factor 1 and IGF binding protein 3 genes and risk of colorectal cancer. Cancer Detect Prev. 2007;31(5):408–16.
Iyengar NM, Hudis CA, Dannenberg AJ. Obesity and inflammation: new insights into breast cancer development and progression. Am Soc Clin Oncol Educ Book. 2013;33:46-51.
Rose DP, Vona-Davis L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr Relat Cancer. 2012;19(6):R225–41.
Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Silva C, Rotellar F, Hernandez-Lizoain JL, Baixauli J, Valenti V, Pardo F, et al. Up-regulation of the novel proinflammatory adipokines lipocalin-2, chitinase-3 like-1 and osteopontin as well as angiogenic-related factors in visceral adipose tissue of patients with colon cancer. J Nutr Biochem. 2011;22(7):634–41.
Liu L, Zhong R, Wei S, Xiang H, Chen J, Xie D, Yin J, Zou L, Sun J, Chen W, et al. The leptin gene family and colorectal cancer: interaction with smoking behavior and family history of cancer. PLoS One. 2013;8(4):e60777.
Creighton CJ, Sada YH, Zhang Y, Tsimelzon A, Wong H, Dave B, Landis MD, Bear HD, Rodriguez A, Chang JC. A gene transcription signature of obesity in breast cancer. Breast Cancer Res Treat. 2012;132(3):993–1000.
Wasserman L, Flatt SW, Natarajan L, Laughlin G, Matusalem M, Faerber S, Rock CL, Barrett-Connor E, Pierce JP. Correlates of obesity in postmenopausal women with breast cancer: comparison of genetic, demographic, disease-related, life history and dietary factors. Int J Obes Relat Metab Disord. 2004;28(1):49–56.
Morimoto LM, Newcomb PA, White E, Bigler J, Potter JD. Insulin-like growth factor polymorphisms and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2005;14(5):1204–11.
Khoury-Shakour S, Gruber SB, Lejbkowicz F, Rennert HS, Raskin L, Pinchev M, Rennert G. Recreational physical activity modifies the association between a common GH1 polymorphism and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17(12):3314–8.
Seti H, Leikin-Frenkel A, Werner H. Effects of omega-3 and omega-6 fatty acids on IGF-I receptor signalling in colorectal cancer cells. Arch Physiol Biochem. 2009;115(3):127–36.
Slattery ML, Lundgreen A, Herrick JS, Caan BJ, Potter JD, Wolff RK. Diet and colorectal cancer: analysis of a candidate pathway using SNPS, haplotypes, and multi-gene assessment. Nutr Cancer. 2011;63(8):1226–34.
McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363(24):2339–50.
Weichhaus M, Broom J, Wahle K, Bermano G. A novel role for insulin resistance in the connection between obesity and postmenopausal breast cancer. Int J Oncol. 2012;41(2):745–52.
Simons CC, van den Brandt PA, Stehouwer CD, van Engeland M, Weijenberg MP. Body size, physical activity, early-life energy restriction, and associations with methylated insulin-like growth factor-binding protein genes in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2014;23(9):1852–62.
Slattery ML, Murtaugh M, Caan B, Ma KN, Neuhausen S, Samowitz W. Energy balance, insulin-related genes and risk of colon and rectal cancer. Int J Cancer. 2005;115(1):148–54.
The Women's Health Initiative Study Group. Design of the Women's Health Initiative clinical trial and observational study. The Women's Health Initiative study group. Control Clin Trials. 1998;19(1):61–109.
WHI Harmonized and Imputed GWAS Data. dbGaP Study Accession: phs000746.v1.p3 [http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000746.v1.p3].
National Cancer Institute. SEER Program: Comparative Staging Guide For Cancer. 1993.
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–16.
Ingelsson E, Langenberg C, Hivert MF, Prokopenko I, Lyssenko V, Dupuis J, Magi R, Sharp S, Jackson AU, Assimes TL, et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic loci regulating glucose and insulin metabolism in humans. Diabetes. 2010;59(5):1266–75.
Nettleton JA, Hivert MF, Lemaitre RN, McKeown NM, Mozaffarian D, Tanaka T, Wojczynski MK, Hruby A, Djousse L, Ngwa JS, et al. Meta-analysis investigating associations between healthy diet and fasting glucose and insulin levels and modification by loci associated with glucose homeostasis in data from 15 cohorts. Am J Epidemiol. 2013;177(2):103–15.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–9.
MacKinnon DP, Fairchild AJ, Fritz MS. Mediation analysis. Annu Rev Psychol. 2007;58:593–614.
Mackinnon DP, Warsi G, Dwyer JH. A simulation study of mediated effect measures. Multivar Behav Res. 1995;30(1):41.
Shrout PE, Bolger N. Mediation in experimental and nonexperimental studies: new procedures and recommendations. Psychol Methods. 2002;7(4):422–45.
Buettner R, Scholmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring). 2007;15(4):798–808.
Institute EB. 1000 genomes browser orientation. In: Based on Project Phase I Data. 2011.
Guo T, Chen T, Gu C, Li B, Xu C. Genetic and molecular analyses reveal G6PC as a key element connecting glucose metabolism and cell cycle control in ovarian cancer. Tumour Biol. 2015;36(10):7649–58.
Abbadi S, Rodarte JJ, Abutaleb A, Lavell E, Smith CL, Ruff W, Schiller J, Olivi A, Levchenko A, Guerrero-Cazares H, et al. Glucose-6-phosphatase is a key metabolic regulator of glioblastoma invasion. Mol Cancer Res. 2014;12(11):1547–59.
Wang B, Hsu SH, Frankel W, Ghoshal K, Jacob ST. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology. 2012;56(1):186–97.
Nettleton JA, McKeown NM, Kanoni S, Lemaitre RN, Hivert MF, Ngwa J, van Rooij FJ, Sonestedt E, Wojczynski MK, Ye Z, et al. Interactions of dietary whole-grain intake with fasting glucose- and insulin-related genetic loci in individuals of European descent: a meta-analysis of 14 cohort studies. Diabetes Care. 2010;33(12):2684–91.
Murad AS, Smith GD, Lewis SJ, Cox A, Donovan JL, Neal DE, Hamdy FC, Martin RM. A polymorphism in the glucokinase gene that raises plasma fasting glucose, rs1799884, is associated with diabetes mellitus and prostate cancer: findings from a population-based, case-control study (the ProtecT study). Int J Mol Epidemiol Genet. 2010;1(3):175–83.
Dong X, Tang H, Hess KR, Abbruzzese JL, Li D. Glucose metabolism gene polymorphisms and clinical outcome in pancreatic cancer. Cancer. 2011;117(3):480–91.
Mazzoccoli G, Panza A, Valvano MR, Palumbo O, Carella M, Pazienza V, Biscaglia G, Tavano F, Di Sebastiano P, Andriulli A, et al. Clock gene expression levels and relationship with clinical and pathological features in colorectal cancer patients. Chronobiol Int. 2011;28(10):841–51.
Kirchhoff K, Machicao F, Haupt A, Schafer SA, Tschritter O, Staiger H, Stefan N, Haring HU, Fritsche A. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia. 2008;51(4):597–601.
Acknowledgements
N/A
Funding
No specific funding was received for this study.
Availability of data and materials
All datasets on which the conclusions of the manuscript rely have been deposited in publicly available WHI repositories (phs000200.v10.p3).
Authors’ contributions
SJ formulated the research question, designed and conducted data analysis, and wrote the article. ES and JP contributed to the study concept, data analysis and interpretation, and drafting of the article. ZZ contributed to the study concept, research design, data interpretation, and drafting of the article. In addition, all authors reviewed the final manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Each institution obtained human subjects committee approval. All participants provided written informed consent. This study was approved by the ethics committees of each participating clinical center of the WHI and the University of California, Los Angeles.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Effect size of glucose metabolism–relevant SNPs on metabolic biomarkers. Table S1.1.Effect size of glucose metabolism–relevant SNPs on glucose level in the pathway of glucose metabolism genetic variants, glucose metabolism traits, and breast cancer risk, stratified by obesity status and obesity-related factors. Table S1.2. Effect size of glucose metabolism–relevant SNPs on HOMA-IR level in the pathway of glucose metabolism genetic variants, glucose metabolism traits, and breast cancer risk, stratified by obesity status and obesity-related factors. Table S1.3. Effect size of glucose metabolism–relevant SNPs on glucose level in the pathway of glucose metabolism genetic variants, glucose metabolism traits, and CRC risk, stratified by obesity status and obesity-related factors. Table S1.4. Effect size of glucose metabolism–relevant SNPs on HOMA-IR level in the pathway of glucose metabolism genetic variants, glucose metabolism traits, and CRC risk, stratified by obesity status and obesity-related factors. Table S1.5. Effect size of glucose metabolism–relevant SNPs on insulin level in the pathway of glucose metabolism genetic variants, glucose metabolism traits, and breast cancer risk, stratified by obesity status and obesity-related factors. Table S1.6. Effect size of glucose metabolism–relevant SNPs on insulin level in the pathway of glucose metabolism genetic variants, glucose metabolism traits, and CRC risk, stratified by obesity status and obesity-related factors. (DOC 188 kb)
Additional file 2:
Characteristics of participants. Table S2.1. Characteristics of participants, stratified by obesity (measured via waist circumference). Table S2.2. Characteristics of participants, stratified by obesity (measured via w/h ratio). Table S2.3. Characteristics of participants, stratified by physical activity level. Table S2.4. Characteristics of participants, stratified by dietary fat intake. (DOC 387 kb)
Additional file 3:
Allele frequencies of 16 glucose metabolism–relevant SNPs. Table S3.1. Allele frequencies of 16 glucose metabolism–relevant SNPs, stratified by obesity (measured via BMI). Table S3.2. Allele frequencies of 16 glucose metabolism–relevant SNPs, stratified by obesity (measured via waist circumference). Table S3.3. Allele frequencies of 16 glucose metabolism–relevant SNPs, stratified by obesity (measured via waist/hip). Table S3.4. Allele frequencies of 16 glucose metabolism–relevant SNPs, stratified by physical activity level. Table S3.5. Allele frequencies of 16 glucose metabolism–relevant SNPs, stratified by dietary fat intake. (DOC 174 kb)
Additional file 4:
Mediation effect of insulin on the relationship between glucose metabolism–relevant SNPs and cancer risk. Table S4.1. Mediation effect of insulin on the relationship between glucose metabolism–relevant SNPs and breast cancer risk, stratified by obesity status and obesity-related factors. Table S4.2. Mediation effect of insulin on the relationship between glucose metabolism–relevant SNPs and CRC risk, stratified by obesity status and obesity-related factors. (DOC 139 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jung, S.Y., Sobel, E.M., Papp, J.C. et al. Effect of genetic variants and traits related to glucose metabolism and their interaction with obesity on breast and colorectal cancer risk among postmenopausal women. BMC Cancer 17, 290 (2017). https://doi.org/10.1186/s12885-017-3284-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-017-3284-7