
Abstract
Background
Vanishing white matter disease (VWMD) is one of the most prevalent inherited leukoencephalopathies, which generally presents in childhood as a progressive disorder while less beginning in adulthood. The present report describes the clinical, neuroimaging, and genetic findings of a female patient with adult-onset VWMD. In addition, to provide a clearer delineation of the clinical and genetic characteristics of female adult-onset VWMD patients, 32 genetically confirmed female adult-onset EIF2B-mutated cases are summarized.
Case presentation
The patient described here suffered from long-term menometrorrhagia prior to manifesting progressive neurological impairments that included tremors, bilateral pyramidal tract injury, cerebellar ataxia, and dementia. To the best of our knowledge, this is the first female patient with adult-onset VWMD suffering from long-term menometrorrhagia attributed to the c.254 T > A and c.496A > G mutations in the EIF2B2 gene; the c.496A > G mutation has not been reported in previous studies. The patient also exhibited metabolic dysfunction. The present findings widen the spectrum of phenotypic heterogeneity observed in VWMD patients.
Conclusions
The present report summarizes 33 female patients with adult-onset VWMD to provide an overview of the clinical and genetic characteristics of this disorder and ovarioleukodystrophy. The mean age of clinical onset in female patients with adult-onset VWMD was 36.8 years and the neurological symptoms primarily included motor and cognitive dysfunction such as paraparesis, cerebellar ataxia, and executive deficits. In addition, ovarian failure occurred in all of these female patients and usually preceded the neurological symptoms. Furthermore, several patients also suffered from metabolic dysfunction. All 33 patients had mutations on EIF2B1–5, and of these, the c.338 G > A mutation in the EIF2B5 gene (p.Arg113His) was the most common. These findings suggest that clinicians should be aware of adult-onset forms of VWMD as well as its typical magnetic resonance imaging (MRI) and clinical characteristics although this pathology is usually recognized as a pediatric disorder. No curative treatment is presently available, and thus early recognition is important to prevent triggering events and to allow for genetic counseling.
Similar content being viewed by others


Background
Vanishing white matter disease (VWMD; OMIM 603896), which is also known as childhood ataxia with central nervous system hypomyelination (CACH), is an autosomal recessive leukoencephalopathy caused by mutations in the EIF2B1–5 genes that encode the subunits of eukaryotic translation initiation factor 2B (eIF2B) [1, 2]. eIF2B is indispensable for the initiation of translation and the regulation of protein synthesis under various conditions, including cell stress [3]. Characteristic neuropathological abnormalities in VWMD patients indicate that there are selective disruptions of oligodendrocytes and astrocytes whereas neurons are spared [4].
Although VWMD was initially recognized as a disorder of young children, it has become clear that there is extreme variation in disease phenotype and severity [5]. VWMD phenotypes range from a congenital form to an infantile form (onset age: 1 year) [6], an early childhood-onset form (onset age: 2–4 years) [5], a juvenile-onset form (onset age: 5–15 years) [7], and an adult-onset form (onset age: > 15 years) [8]. The severe forms of VWMD begin in the prenatal or early infantile period and manifest as cerebellar ataxia and spasticity and lead to early death [9]. Milder variants begin in adolescence or adulthood and are characterized by a slow disease progression [10]. The clinical signs in adults include seizures, spasticity, cerebellar syndrome, dementia, and manifestations of ovarian failure. Adult-onset VWMD occurring in association with primary ovarian failure is described as ovarioleukodystrophy [11], which is an extremely rare condition. Since it was first identified in 1996 [12], fewer than 30 genetically confirmed cases of ovarioleukodystrophy have been reported in the literature. Due to the small number of known cases, most clinicians have a relative lack of knowledge regarding the clinical presentation and neuroimaging features of VWMD ovarioleukodystrophy. Furthermore, there are many phenotypic variants of VWMD, which makes a definitive diagnosis difficult.
The present report describes a female patient with adult-onset VWMD who had two novel heterozygous mutations in the EIF2B2 gene. Distinct from other female cases that are frequently associated with premature ovarian failure, this is the first female patient with adult-onset VWMD suffering from long-term menometrorrhagia. To provide a clearer delineation of the clinical evaluation and genetic presentation of female patients with this disease, the relevant literature was reviewed. This report describes the wide range of phenotypic variants of VWMD and provides insight into early diagnostic strategies.
Case presentation
A 25-year-old female presented with a 6-year history of long menstrual periods and a 4-year history of tremors in both hands. She showed clear signs of developmentally delayed intelligence in childhood. The neurological examination revealed a positive Babinski sign in the right lower limb and a slight decline in intellect (Wechsler Adult Intelligence Scale IQ score, IQ = 81), with particularly decreased cognitive scores on calculation and memory (Mini-mental State Examination, [MMSE] = 24/30). According to the physical examination, the patient was overweight (BMI 27.3 kg/m2), but no other abnormalities were found, including with regard to blood pressure, temperature, and pulse rate. She often felt tired and slept (cumulative sleep time 10–12 h/day). She had a slightly increased appetite, leading to four meals per day plus snacks. Her hands and feet sweat even in normal temperature. No similar symptoms were found in the patient’s relatives, and no head trauma or stress was reported.
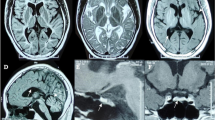
She was first admitted as a gynecological outpatient for prolonged menstruation and subsequently transferred to our department due to an abnormal brain magnetic resonance image (MRI), which revealed diffuse and symmetric white matter abnormalities, with lesions having cerebrospinal fluid (CSF)-like signals (Fig. 1a). 18Flurodeoxyglucose positron emission tomography/computed tomography revealed decreased metabolism in the left frontal, parietal, and temporal lobes as well as in the cerebellum, and this may have been correlated with patient’s impaired calculation and memory deficits (Fig. 1b). Brain diffusion tensor imaging disclosed reduced fibrous white bundles among bilateral periventricular areas (Fig. 1c). Initial blood chemistry revealed only hypochromic microcytic anemia (hemoglobin, 82 g/L) and mild hypertriglyceridemia (triglycerides, 2.35 mmol/L) and hypoglycemia (glucose, 3.78 mmol/L). A cerebral spinal fluid (CSF) analyses were negative for encephalitis and paraneoplastic autoimmune encephalitis, and the patient had normal levels of myelin basic protein and aquaporin-4 with no neuromyelitis optica and a normal cell count without oligoclonal bands. A gynecological examination revealed normal levels of gonadal hormones, and an ultrasound revealed normal ovaries and uterus. A pathological examination of the endometrium showed both simple and complex hyperplasia based on a curettage surgery due to long duration menstruation and a large volume of menstrual blood in the patient.

Neuroimaging results of the VWMD patient. a Axial T1-weighted, T2-weighted, Flair, DWI, ADC and coronal T1-weighted images showing extensive cerebral white matter abnormalities with central areas resembling signal intensity of cerebrospinal fluid surrounded by a rim of hyperintensity in the periventricular and subcortical regions. White-matter rarefaction and cystic degeneration are more evident in subcortical and periventricular regions. Obvious brain atrophy was also detected. b Brain 18F-FDG PET showed decreased FDG uptake in left frontal, parietal and temporal lobe. Compared with right side, the decline rate is 17, 12 and 17%. c Brain DTI disclosed fibrous white bundle was reduced sparsely among bilateral periventricular
Targeted sequencing performed on proband to screen pathogenic mutations. PCR combined Sanger sequencing then performed on three family members including one affected and two unaffected individuals to determine whether mutations cosegregate with disease. Blood samples were collected and stored in 4 °C until analysis. DNA isolation and high-throughput sequencing Genomic DNA was extracted from peripheral blood according to manufacturer’s instructions (QIAGEN, Hilden, Germany). DNA sequencing libraries were then prepared as followed according to Illumina standard protocol: genomic DNA was fragmented; Illumina adapters were ligated to the fragments after ‘A’ ligating to their 3’ends; Fragments with sample size in 200 to 500 base pair were selected and amplified by PCR (each sample is tagged with a unique index during this procedure). Fragments in the exonic regions of targeted genes were captured by a specific Hereditary Leukoencephalopathies Disease GenePanel using biotinylatedoligo-probes (MyGenostics GenCap Enrichment Technologies, MyGenostics, Baltimore, MD, USA). The Panel was designed to detect the coding region of 165 genes which cover almost all of genes that reported to relate to hereditary leukoencephalopathies disease (Additional file 1: Table S1). The capture experiment was conducted according to the manufacturer’s protocol. Briefly, 1 μg DNA library was mixed with Buffer BL and GenCap gene panel probe. The mixture was heated at 95 °C for 7 min and 65 °C for 2 min. Adding 23 μl of the 65 °C prewarmed Buffer HY (MyGenostics) to mixture, then hybridizate at 65 °C for 22 h. After adding 64 μl 2X binding buffer and 80 μl MyOnebeads (Life Technology), the hybrid mixture was transferred to the tube. The mixture was rotated and the beads were washed. The bound DNA was then eluted followed by amplification activated at 98 °C for 30 s (1 cycle), 98 °C for 25 s, 65 °C for 30s, 72 °C for 30s (15 cycles), 72 °C for 5 min (1 cycle). The PCR products were purified and then sequenced by Illumina HiSeq Tm 2000 sequencer, generating 2 × 100 bp reads. Base was called using the Off-Line Base Caller v1.9.
We diagnosed vanishing white matter disease (VWMD) based on compound heterozygosis for the EIF2B2 gene mutation and the diffuse symmetric abnormality in white matter Fig. 2.

Genetic results of the VWMD patient. Genomic sequence chromatograms of the missense in EIF2B2 in patient’s family members: c.254 T > A and c.496A > G
A 10-month follow-up examination showed a mild decline in intelligence (IQ decreased from 81 to 73), a slightly aggravated pyramidal tract (positive bilateral Babinski and Chaddock signs), and light cerebellar ataxia (unsteady bilateral knee-shin and finger-nose signs). The tremors in the hands had not changed, and she still demonstrated a prolonged menstrual period with an irregular menstrual cycle 3 months after the curettage. A brain MRI showed an image coincident with the prior one. Written informed consent was obtained from the patient prior to publication of this report.
Discussion and conclusions
Review of the literature
Prior to this report, 32 cases of female patients with adult-onset VWMD had been published. To provide an overview of the clinical characteristics of female patients with adult-onset VWMD and ovarioleukodystrophy, searches of the PubMed and MEDLINE databases were performed in March 2019 using the terms “adult onset vanishing white matter disease,” “late onset vanishing white matter disease,” “eukaryotic translation initiation factor 2B,” or “ovarioleukodystrophy”; all identified articles published in English and articles referenced therein were reviewed. The patient inclusion criteria consisted of: disease onset after the age of 15 years, mutation in one of the EIF2B1–5 genes, and being female.
In addition to the present case, a total of 32 genetically confirmed cases were identified and summarized to provide an overview of the clinical and genetic features of female adult-onset VWMD (Table 1).
Neurological evaluation
The mean age of onset of female patients with adult-onset VWMD was 36.8 years (range: 16–66 years, standard deviation [SD]: 14.3). Motor dysfunction was detected in 29 patients (87.8%) and gait instability, spastic paraparesis, and cerebellar ataxia were the three main VWMD symptoms. More specifically, of the 33 patients, 11 (30%) suffered from spastic paraparesis, 9 (27.2%) exhibited gait instability, and 6 (18.1%) showed cerebellar ataxia. Tremors and weakness of the limbs were also evident in the patients.
Of the 33 patients, 25 (75.8%) had available data about cognitive function. Cognition was impaired in 20 patients (80%; mean MMSE score: 19.5, range: 4–27, SD: 6.89) and executive and memory deficits were the major signs of cognitive dysfunction. IQ tests (mean score: 65, SD: 11.2) revealed declines in intellect in eight patients.
Seizures occurred in 10 patients (30.3%) and paresthesias and psychiatric symptoms also caused deterioration in the VWMD patients.
Evaluation of ovarian function
Of the 33 patients, 24 (72.7%) exhibited ovarian failure, 13 (54.2%) had secondary amenorrhea (mean onset age: 27.8 years, range: 21–37, SD: 5.73), 7 (29.2%) suffered primary amenorrhea, 3 (9.1%) presented with delayed menarche, and 1 suffered infertility; ovarian failure always preceded the neurological symptoms. In contrast to all previously identified cases, the present patient was the first to report suffering from long-term menometrorrhagia.
Evaluation of metabolic dysfunction
Of the 33 patients in the present report, 5 (15.1%) were obese, 6 (18.2%) exhibited hyperlipemia, and 2 showed changes in glucose metabolism that presented as hypoglycemia.
Genetic findings
All of the female VWMD patients included in the present report had their diagnoses genetically confirmed: 18 (54.5%) exhibited mutations in the EIF2B5 gene, 7 (21.2%) had them in the EIF2B2 gene, and 4 (12.1%) had them in the EIF2B4 gene. The recurrent c.338 G > A mutation in the EIF2B5 gene (p.Arg113His) was found in 12 patients (36.4%), including in 11 patients in a homozygous state. In the only case of a compound heterozygote for this recurrent mutation, the second mutation was the c.538 C > T. EIF2B2 gene, which is the second most common gene mutation in VWMD patients. The patient described in the present report was also confirmed to be carrying a compound heterozygote for the EIF2B2 gene mutations: c.254 T > A and c.496A > G. The c.254 T > A mutation in the EIF2B2 gene has been reported in female patients with adult-onset VWMD but, to the best of our knowledge, the c.496A > G missense mutation in the EIF2B2 gene has never been reported. All reported cases of the EIF2B4 mutations were heterozygous. Mutations in the EIF2B3 and EIF2B1 genes are relatively rare and only one patient was confirmed to be carrying an EIF2B1 gene mutation.
The present findings indicate that VWMD presents along a wider clinical spectrum than previously thought. The mean age of clinical onset of the female adult-onset VWMD patients in the present report was 36.8 years old and the related neurological symptoms primarily involved motor and cognitive dysfunction such as paraparesis, cerebellar ataxia, and executive deficits. Ovarian failure occurred in all female VWMD patients and usually preceded the neurological symptoms. In addition, several patients suffered from physical dysfunction. All of the female adult-onset VWMD patients had mutations on EIF2B1–5, and of these, the c.338 G > A mutation in the EIF2B5 gene (p.Arg113His) was the most common.
Discussion
VWMD generally presents in childhood as a progressive disorder with episodes of rapid and major neurological deterioration induced by stressors [29]. Much milder variants begin in adulthood and are characterized by slowly progressive spastic paraparesis and cerebellar ataxia. Ovarian dysfunction is frequently reported in females with VWMD, particularly those with abnormal menstruation, and can precede the neurological symptoms [30]. This condition is known as ovarioleukodystrophy, which is an extremely rare condition as fewer than 30 genetically confirmed cases have been reported. The present report summarized genetically confirmed cases in the literature to provide an overview of the clinical and genetic features of adult-onset female VWMD. To the best of our knowledge, this case series is the largest ever reported and one of the first to describe a female adult-onset VWMD patient suffering from progressive neurological impairment, including tremors, bilateral pyramidal tract injury, cerebellar ataxia, and dementia. Prior to the appearance of these neurological symptoms, this patient showed long-term menometrorrhagia. This case was distinctly different from previous cases, as the ovarian function and morphological structure of our patient were normal. The present patient also exhibited physical symptoms due to systemic injuries that included metabolic dysfunction (obesity, hypertriglyceridemia, and hypoglycemia), abnormal feeding behaviors (more frequent eating), and autonomic nervous dysfunction (sweating of the limbs). Based on these clinical manifestations, it is possible that multi-system injuries were involved in the pathogenesis of this case of VWMD.
VWMD is an autosomal recessive leukoencephalopathy caused by mutations in any of five genes (EIF2B1–5) encoding the eIF2B subunits, which is a protein complex involved in the regulation of the first step of protein synthesis [31]. Activity of ElF2B guanine nucleotide exchange factor is subject to direct control through its own phosphorylation, which leads to a rapid reduction in protein synthesis [32]. Neuropathological abnormalities in patients with VWMD indicate selective disruption of oligodendrocytes and astrocytes with sparing of neurons. However, how the gene mutation causes nerve injury is unknown. Until now, more than 120 mutations in EIF2B1–5 have been identified [33]. Our patient was confirmed to carry a compound heterozygote for the EIF2B2 gene mutations c.254 T > A and c.496A > G. The c.254 T > A mutation in the EIF2B2 gene has been presented three times (Table 2) [16, 34], including one female adult-onset female with VWMD [35]. The c.254 T > A missense mutation leads to a single substitution within coding region from thymine to adenine, resulting from valine to glutamic acid (p.V85E). The function of mutant c.254 T > A was predicted to be pathological and had been reported in the Human Gene Mutation Database (HGMD) database (http://www.hgmd.cf.ac.uk/ac/index.php). In 2011, Matsukawa revealed that the mutant c.254 T > A subunit could cause approximately 20% decreased GDP/GTP exchange activity of eIF2B [16]. To the best of our knowledge, the c.496A > G missense mutation in the EIF2B2 gene has never been reported. It causes adenine change to guanine, resulting from methionine to valine (p.M166 V). This mutation does not belong to polymorphism sites and has never been found in the population. SIFT (http://sift.jcvi.org/) and POLYPHEN2 (http://genetics.bwh.harvard.edu/pph2/) were used to analyze the amino acid substitutions of p.M166 V (Fig. 3a and b). Both programs predicted the mutation c.496A > G to be deleterious, which means they probably damage and affect protein functions. Furthermore, protein tertiary structures were modeled with SWISS-MODEL (http://www.swissmodel.expasy.or-g/), which predict the sequence homology. The p.V85E protein model revealed that the mutation c.254 T > A lead to the amino acid side chain change through the substitution of valine to glutamic acid. The p.M166 V protein model predicted that the mutation c.496A > G affected the amino acid side chain by the substitution of methionine to valine (Fig. 3c).

Function prediction and protein molecular models: a SIFT and b POLYPHEN2 online websites were used to analyze the amino acid substitutions of p.M166 V. Both programs predicted the mutation c.496A > G to be deleterious, which means they probably damage and affect protein functions. c The mutation protein E85 has a different side chain to the wild-type protein V85. The wide-type protein has a longer side chain than the mutation V166 protein
Brain MRI scans of VWMD patients typically show diffuse symmetric abnormalities of the cerebral white matter with the lesions exhibiting characteristic cerebrospinal fluid (CSF)-like signals [36]. These white matter hyperintensities are followed by cerebral white matter rarefaction and cystic degeneration in which the white matter is replaced by fluid [37].
Conclusions
In conclusion, the present report described the clinical, neuroimaging, and genetic findings of a female adult-onset patient with VWMD. To the best of our knowledge, this is the first female adult-onset patient with VWMD suffering from long-term menometrorrhagia due to novel EIF2B2 gene mutations. Furthermore, the cases of 33 female patients with adult-onset VWMD were summarized to provide an overview of the clinical and genetic characteristics of this disorder and ovarioleukodystrophy. Our description enlarges the spectrum of phenotypic heterogeneity in VWMD. Clinicians should be aware of adult and milder forms of VWMD, although VWMD is usually recognized as a pediatric disorder. No curative treatment is currently available. Thus, early recognition is important to prevent triggering events and to allow for genetic counseling.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- CACH:
-
Central nervous system hypomyelination
- CSF:
-
Cerebrospinal fluid
- eIF2B:
-
eukaryotic translation initiation factor 2B
- IQ:
-
Wechsler Adult Intelligence Scale IQ score
- MMSE:
-
Mini-mental State Examination
- MRI:
-
Magnetic resonance image
- SD:
-
Standard Deviation
- VWMD:
-
Vanishing white matter disease
References
Schiffmann R, Fogli A, van der Knaap MS, Boespflug-Tanguy O. Childhood Ataxia with Central Nervous System Hypomyelination/Vanishing White Matter. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, Amemiya A, editors. Seattle: GeneReviews®; 1993.
Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS. Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol. 2010;69(10):987–96.
Pronk JC, van Kollenburg B, Scheper GC, van der Knaap MS. Vanishing white matter disease: a review with focus on its genetics. Ment Retard Dev Disabil Res Rev. 2006;12(2):123–8.
van der Knaap MS, Barth PG, Gabreels FJ, Franzoni E, Begeer JH, Stroink H, Rotteveel JJ, Valk J. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48(4):845–55.
Schiffmann R, Moller JR, Trapp BD, Shih HH, Farrer RG, Katz DA, Alger JR, Parker CC, Hauer PE, Kaneski CR, et al. Childhood ataxia with diffuse central nervous system hypomyelination. Ann Neurol. 1994;35(3):331–40.
Yavuz H. A review of infantile vanishing white matter disease and a new mutation. Acta Neurol Taiwanica. 2017;26(4):167–76.
van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol. 2006;5(5):413–23.
van der Lei HD, van Berkel CG, van Wieringen WN, Brenner C, Feigenbaum A, Mercimek-Mahmutoglu S, Philippart M, Tatli B, Wassmer E, Scheper GC, et al. Genotype-phenotype correlation in vanishing white matter disease. Neurology. 2010;75(17):1555–9.
van der Knaap MS, van Berkel CG, Herms J, van Coster R, Baethmann M, Naidu S, Boltshauser E, Willemsen MA, Plecko B, Hoffmann GF, et al. eIF2B-related disorders: antenatal onset and involvement of multiple organs. Am J Hum Genet. 2003;73(5):1199–207.
Riecker A, Nagele T, Henneke M, Schols L. Late onset vanishing white matter disease. J Neurol. 2007;254(4):544–5.
Fogli A, Rodriguez D, Eymard-Pierre E, Bouhour F, Labauge P, Meaney BF, Zeesman S, Kaneski CR, Schiffmann R, Boespflug-Tanguy O. Ovarian failure related to eukaryotic initiation factor 2B mutations. Am J Hum Genet. 2003;72(6):1544–50.
Schiffmann R, Tedeschi G, Kinkel RP, Trapp BD, Frank JA, Kaneski CR, Brady RO, Barton NW, Nelson L, Yanovski JA. Leukodystrophy in patients with ovarian dysgenesis. Ann Neurol. 1997;41(5):654–61.
Lee HN, Koh SH, Lee KY, Ki CS, Lee YJ. Late-onset vanishing white matter disease with compound heterozygous EIF2B5 gene mutations. Eur J Neurol. 2009;16(3):e42–3.
La Piana R, Vanderver A, van der Knaap M, Roux L, Tampieri D, Brais B, Bernard G. Adult-onset vanishing white matter disease due to a novel EIF2B3 mutation. Arch Neurol. 2012;69(6):765–8.
Robinson ME, Rossignol E, Brais B, Rouleau G, Arbour JF, Bernard G. Vanishing white matter disease in French-Canadian patients from Quebec. Pediatr Neurol. 2014;51(2):225–32.
Matsukawa T, Wang X, Liu R, Wortham NC, Onuki Y, Kubota A, Hida A, Kowa H, Fukuda Y, Ishiura H, et al. Adult-onset leukoencephalopathies with vanishing white matter with novel missense mutations in EIF2B2, EIF2B3, and EIF2B5. Neurogenetics. 2011;12(3):259–61.
Sambati L, Agati R, Bacci A, Bianchi S, Capellari S. Vanishing white matter disease: an Italian case with A638G mutation in exon 5 of EIF2B2 gene, an unusual early onset and a long course. Neurol Sci. 2013;34(7):1235–8.
Ghezzi L, Scarpini E, Rango M, Arighi A, Bassi MT, Tenderini E, De Riz M, Jacini F, Fumagalli GG, Pietroboni AM, et al. A 66-year-old patient with vanishing white matter disease due to the p.Ala87Val EIF2B3 mutation. Neurology. 2012;79(20):2077–8.
Shimada S, Shimojima K, Sangu N, Hoshino A, Hachiya Y, Ohto T, Hashi Y, Nishida K, Mitani M, Kinjo S, et al. Mutations in the genes encoding eukaryotic translation initiation factor 2B in Japanese patients with vanishing white matter disease. Brain and Development. 2015;37(10):960–6.
Woody AL, Hsieh DT, McIver HK, Thomas LP, Rohena L. Infantile onset vanishing white matter disease associated with a novel EIF2B5 variant, remarkably long life span, severe epilepsy, and hypopituitarism. Am J Med Genet A. 2015;167A(4):826–30.
Ohtake H, Shimohata T, Terajima K, Kimura T, Jo R, Kaseda R, Iizuka O, Takano M, Akaiwa Y, Goto H, et al. Adult-onset leukoencephalopathy with vanishing white matter with a missense mutation in EIF2B5. Neurology. 2004;62(9):1601–3.
Labauge P, Horzinski L, Ayrignac X, Blanc P, Vukusic S, Rodriguez D, Mauguiere F, Peter L, Goizet C, Bouhour F, et al. Natural history of adult-onset eIF2B-related disorders: a multi-centric survey of 16 cases. Brain. 2009;132(Pt 8):2161–9.
Carra-Dalliere C, Horzinski L, Ayrignac X, Vukusic S, Rodriguez D, Mauguiere F, Peter L, Goizet C, Bouhour F, Denier C, et al. Natural history of adult-onset eIF2B-related disorders: a multicentric survey of 24 cases. Rev Neurol (Paris). 2011;167(11):802–11.
Denier C, Orgibet A, Roffi F, Jouvent E, Buhl C, Niel F, Boespflug-Tanguy O, Said G, Ducreux D. Adult-onset vanishing white matter leukoencephalopathy presenting as psychosis. Neurology. 2007;68(18):1538–9.
Peter L, Niel F, Catenoix H, Jung J, Demarquay G, Petiot P, Rudigoz RC, Boespflug-Tanguy O, Ryvlin P, Mauguiere F. Acute neurological deterioration in ovarioleukodystrophy related to EIF2B mutations: pregnancy with oocyte donation is a potentially precipitating factor. Eur J Neurol. 2008;15(1):94–7.
Ibitoye RT, Renowden SA, Faulkner HJ, Scolding NJ, Rice CM. Ovarioleukodystrophy due to EIF2B5 mutations. Pract Neurol. 2016;16(6):496–9.
Takano K, Tsuyusaki Y, Sato M, Takagi M, Anzai R, Okuda M, Iai M, Yamashita S, Okabe T, Aida N, et al. A Japanese girl with an early-infantile onset vanishing white matter disease resembling Cree leukoencephalopathy. Brain and Development. 2015;37(6):638–42.
Humayun M, Khan A. Case Report: Cerebral leukodystrophy and the gonadal endocrinopathy: a rare but real association. F1000Res. 2018;7:158.
Turon-Vinas E, Pineda M, Cusi V, Lopez-Laso E, Del Pozo RL, Gutierrez-Solana LG, Moreno DC, Sierra-Corcoles C, Olabarrieta-Hoyos N, Madruga-Garrido M, et al. Vanishing white matter disease in a spanish population. J Cent Nerv Syst Dis. 2014;6:59–68.
Scali O, Di Perri C, Federico A. The spectrum of mutations for the diagnosis of vanishing white matter disease. Neurol Sci. 2006;27(4):271–7.
Fogli A, Boespflug-Tanguy O. The large spectrum of eIF2B-related diseases. Biochem Soc Trans. 2006;34(Pt 1):22–9.
van der Knaap MS, Leegwater PA, Konst AA, Visser A, Naidu S, Oudejans CB, Schutgens RB, Pronk JC. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol. 2002;51(2):264–70.
Maletkovic J, Schiffmann R, Gorospe JR, Gordon ES, Mintz M, Hoffman EP, Alper G, Lynch DR, Singhal BS, Harding C, et al. Genetic and clinical heterogeneity in eIF2B-related disorder. J Child Neurol. 2008;23(2):205–15.
Wu Y, Pan Y, Du L, Wang J, Gu Q, Gao Z, Li J, Leng X, Qin J, Wu X, et al. Identification of novel EIF2B mutations in Chinese patients with vanishing white matter disease. J Hum Genet. 2009;54(2):74–7.
Unal O, Ozgen B, Orhan D, Tokatli A, Hismi BO, Dursun A, Coskun T, Kalkanoglu-Sivri HS. Vanishing white matter with hepatomegaly and hypertriglyceridemia attacks. J Child Neurol. 2013;28(11):1509–12.
Hanefeld F, Holzbach U, Kruse B, Wilichowski E, Christen HJ, Frahm J. Diffuse white matter disease in three children: an encephalopathy with unique features on magnetic resonance imaging and proton magnetic resonance spectroscopy. Neuropediatrics. 1993;24(5):244–8.
Fogli A, Schiffmann R, Bertini E, Ughetto S, Combes P, Eymard-Pierre E, Kaneski CR, Pineda M, Troncoso M, Uziel G, et al. The effect of genotype on the natural history of eIF2B-related leukodystrophies. Neurology. 2004;62(9):1509–17.
Acknowledgements
Not applicable
Funding
This study was supported by Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (No.ZYLX201837) and National Key R&D Program of China (No. 2017YFC1310103). These funders provided financial support for this report.
Author information
Authors and Affiliations
Contributions
CBW examined the patient and acquire of data. CBW and QQ drafted the manuscript. JPJ revised the article. FC and AHZ performed and analyzed the endometrium biopsy. FW, XMZ and QQ analyzed the mutation. RC and JHL participated in the design of the case report and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The institutional review board of Xuan Wu Hospital affiliated to Capital Medical University approved the study.
Consent for publication
Written informed consent was obtained from the parents of the patient for publication of this case report and accompanying brain MRI images and mutation results. A copy of consent to publish is available.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Genes detected relate to hereditary leukoencephalopathies disease. (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wei, C., Qin, Q., Chen, F. et al. Adult-onset vanishing white matter disease with the EIF2B2 gene mutation presenting as menometrorrhagia. BMC Neurol 19, 203 (2019). https://doi.org/10.1186/s12883-019-1429-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-019-1429-9