
Abstract
Background
Autosomal recessive Myotonia congenita (Becker’s disease) is caused by mutations in the CLCN1 gene. The condition is characterized by muscle stiffness during sustained muscle contraction and variable degree of muscle weakness that tends to improve with repeated contractions.
Case presentation
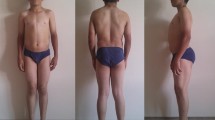
A 21-year-old man presented with transient muscle stiffness since the last 10 years. He had difficulty in initiating movement and experienced muscle weakness after rest, which typically improved after repeated contraction (warm-up phenomenon). There was no significant family history. Medical examination showed generalized muscle hypertrophy. Serum creatine kinase level was 2-fold higher than the normal value. Electromyogram showed myotonic discharges. DNA sequence analysis identified a novel splice mutation (c.1401 + 1G > A) and a known mutation (c.1657A > T,p.Ile553Phe). He rapidly responded to treatment with mexiletine 100 mg three times a day for 6 months.
Conclusions
This case report of autosomal recessive Myotonia congenita caused by a novel compound heterozygous mutation expands the genotypic spectrum of CLCN1 gene.
Similar content being viewed by others


Background
Autosomal recessive Myotonia congenita (Becker’s disease) is caused by mutations in CLCN1 gene, which is located on the 7q35 chromosomal region and contains 23 coding exons. The condition is characterized by muscle stiffness during sustained muscle contraction and a variable degree of muscle weakness, which tends to improve with repeated contraction. The CLCN1 gene is responsible for chloride (Cl−) conductance in skeletal muscles, which contributes significantly to the repolarization of action potential.
Till date, several mutations of the CLCN1 gene such as missense and nonsense mutations have been reported in different populations across the world. To the best of our knowledge, only a few splicing mutations of the CLCN1 gene have been reported. These mutations may induce depolarisation of the skeletal muscle cell membrane which leads to hyperexcitablity (myotonia), which may improve on treatment with sodium (Na+) blocking agents such as mexiletine [1,2,3,4,5]. In this study, we report a Chinese patient suffering from recessive Myotonia congenita caused by compound heterozygous mutations which includes a novel splicing mutation.
Case presentation
A 21-year-old man complained of transient muscle stiffness since 10 years. He experienced difficulty in initiating movement and felt muscle weakness after rest. However the symptoms typically improved after repeated contraction (warm-up phenomenon). The symptoms tended to aggravate during cold weather. He was unable to open his eyes immediately after washing his face with cold water. He had non-consanguineous parents and there was no significant family history. Further, the patient showed normal growth and development. Medical examination showed generalized muscle hypertrophy and normal muscle strength as assessed with the Medical Research Council (MRC) sum score. Deep tendon reflexes were attenuated. No nerve dysfunction or sensory deficit was noted. The serum creatine kinase level was 2-fold higher than the upper limit of the normal reference level. Electromyogram showed myotonic discharges. Biceps muscle biopsy specimen was obtained after written informed consent of the patient. The specimen was precooled with isopentane and frozen in liquid nitrogen. A section of muscle biopsy specimen was stained with hematoxylin-eosin (HE) and modified Gomori’s trichrome (MGT). Activity of oxidative enzymes such as succinate dehydrogenase (SDH), NADH-tetrazolium reductase (NADH-TR), and cytochrome c oxidase (COX) were normal. Next generation sequencing of the DNA sample of this patient identified a novel splice mutation (c.1401 + 1G > A) (which was inherited from his father) and a known mutation (c.1657A > T, p.Ile553Phe) (which was inherited from his mother) (Fig. 1). The novel splice mutation was not detected in Human Gene Mutation Database or any of the 200 healthy controls. The splicing site, analyzed by Human Splicing Finder, is implicated in the alteration of the wild-type donor site and most probably have an impact on splicing. Mutation Taster software predicted the effects of the mutation to be ‘disease causing’. The patient was prescribed mexiletine 100 mg three times per day for about 6 months. The patient responded well to the treatment and was completely relieved of his symptoms.

Pedigree and mutation analysis of the patient. The c.1401 + 1 G > A mutation (red arrow) and c.1657 A > T mutation (red arrow), inherited from his father (I:1) and mother (I:2) respectively, were found in the patient (II)
Discussion and conclusions
Our patient showed transient muscle stiffness and weakness after rest which improved with repeated contractions. The symptoms were aggravated by cold weather and stress. EMG revealed myotonic discharges and gene sequencing analysis identified novel compound heterozygous mutations in CLCN1 gene. Finally, a diagnosis of recessive myotonia congenita was established.
The CLCN1 gene encodes the major skeletal muscle chloride channel CLCN-1. CLCN-1 plays an important part in the maintenance of resting potential, which is activated during the course of depolarization. The mutations of CLCN1 gene were scattered throughout the entire sequence of the channel protein. The mutations included insertion/deletion, missense, nonsense, and splicing mutations. To the best of our knowledge, very few studies have reported splicing mutations of CLCN1 gene [1, 2, 4, 5]. The sequences associated with splicing mutation contains two splice sites: one is a donor site, which is an invariant GU at the 5′ end of the intron and another is an acceptor site, which is an invariant AG at the 3′ end of the intron. Meyer-Kleine et al... reported a 5′ splice-donor mutation (c.1471 + 1G > A) in three GM family, which can change the highly conserved consensus sequence [4]. Gianna et al reported four cases that harbored splicing mutations(c.563G > T, c.1169-5 T > G, c.1251 + 1G > A and c.1931-2A > G), which led to skipping exons in a frameshift change during RNA translation or premature termination [5]. In our case, the novel splicing mutation c.1401 + 1G > A in intron 12 affected the 5′ splice-donor sequences resulting in exons skipping and generation of out-of-frame mRNA. However, the specific mechanism is yet to be elucidated. Another mutation (c.1657A > T,p.Ile553Phe) has been reported in Chinese GM patients [6], which may induce small changes in channel properties. This indicates that the mutation may be associated with certain ethnic populations. CLCN-1 is a voltage-dependent ion channel that mediates chloride conductance in the skeletal muscle cell membrane. The mutations of CLCN1 gene may result in a decrease in channel opening probability and render the membrane hyperexcitable, which is manifested as myotonia. This can be improved by administration of Na+ blocking agents, such as mexiletine [7].
In conclusion, this case report of autosomal recessive myotonia congenita caused by a novel compound heterozygous mutation expands the genotypic spectrum of CLCN1 gene.
Abbreviations
- COX:
-
cytochrome c oxidase
- GM:
-
generalized myotonia
- HE:
-
hematoxylin-eosin
- MGT:
-
modified Gomori’s trichrome
- MRC:
-
Medical Research Council
- NADH-TR:
-
NADH-tetrazolium reductase
- SDH:
-
succinate dehydrogenase
References
McKay OM, Krishnan AV, Davis M, Kiernan MC. Activity-induced weakness in recessive myotonia congenita with a novel (696+1G>a) mutation. Clin Neurophysiol. 2006;117:2064–8.
Ulzi G, Lecchi M, Sansone V, Redaelli E, Corti E, Saccomanno D, et al. Myotonia congenita: novel mutations in CLCN1 gene and functional characterizations in Italian patients. J Neurol Sci. 2012;318:65–71.
Cheng W, Tian J, Burgunder JM, Hunziker W, Eng HL. Myotonia congenita-associated mutations in chloride channel-1 affect zebrafish body wave swimming kinematics. PLoS One. 2014;9:e103445.
Meyer-Kleine C, Steinmeyer K, Ricker K, Jentsch TJ, Koch MC. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am J Hum Genet. 1995;57:1325–34.
Ulzi G, Sansone VA, Magri F, Corti S, Bresolin N, Comi GP, et al. In vitro analysis of splice site mutations in the CLCN1 gene using the minigene assay. Mol Biol Rep. 2014;41:2865–74.
Burgunder JM, Huifang S, Beguin P, Baur R, Eng CS, Seet RC, et al. Novel chloride channel mutations leading to mild myotonia among ChineseNeuromuscul Disord. 2008;18:633–40.
Andersen G, Hedermann G, Witting N, Duno M, Andersen H, Vissing J. The antimyotonic effect of lamotrigine in non-dystrophic myotonias: a double-blind randomized study. Brain. 2017;140:2295–305.
Acknowledgements
The authors thank Medjaden Bioscience Limited for proofreading the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No, 81601088), Natural Science Foundation of Jilin Province (No, 20160520164JH) and the 7th Youth Foundation of the First Hospital of Jilin University (No, JDYY72016028).
Availability of data and materials
All data are presented in the manuscript. There are no additional data.
Consent to publish
Written informed consent for publication of this Case Report was obtained from the patient. A copy of written consent form is available for review to the Editor of this journal.
Author information
Authors and Affiliations
Contributions
JM drafted the manuscript and figures; XW and XL designed and analyzed the study; ZK and YG analyzed and interpreted histological data; XY revised the manuscript and gave the final approval of the version to be published. All authors read and approved the contents of the case report.
Corresponding author
Ethics declarations
Authors’ information
All of authors are from the Department of Neurology and the Neuroscience Center of the First Affiliated Hospital of Jilin University.
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Miao, J., Wei, Xj., Liu, Xm. et al. A case report: autosomal recessive Myotonia congenita caused by a novel splice mutation (c.1401 + 1G > A) in CLCN1 gene of a Chinese Han patient. BMC Neurol 18, 154 (2018). https://doi.org/10.1186/s12883-018-1153-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-018-1153-x