
Abstract
Background
The etiologic determinants of stroke in young adults remain a diagnostic challenge in up to one-fourth of cases. Increasing evidences led to consider Fabry’s disease (FD) as a possible cause to check up. We aimed at evaluating the prevalence of unrecognized FD in a cohort of patients with juvenile stroke in northern Sardinia.
Methods
For this study, we enrolled 178 patients consecutively admitted to our Neurological Ward for ischemic stroke, transient ischemic attack, intracerebral haemorrhage, neuroradiological evidence of silent infarcts, or white matter lesions possibly related to cerebral vasculopathy at brain MRI, and cerebral venous thrombosis. The qualifying events have to occur between 18 and 55 years of age.
Results
We identified two patients with an α-galactosidase A gene variant, with a prevalence of 0.9 %. According to recent diagnostic criteria, one patient, included for the occurrence of multiple white matter lesions at brain MRI, had a diagnosis of definite FD, the other, included for ischemic stroke, had a diagnosis of uncertain FD.
Conclusions
Our study places in a middle position among studies that found a prevalence of FD up to 4 % and others that did not find any FD patients. Our findings confirm that FD should be considered in the differential diagnosis of patients with juvenile stroke, particularly those with a personal or familial history positive for cerebrovascular events, or evidence of combined cardiologic and/or renal impairment. All types of cerebrovascular disorders should be screened for FD, including patients with white matter lesions possibly related to cerebral vasculopathy at brain MRI.
Similar content being viewed by others


Background
Stroke is the first leading cause of disability in adults and the fourth cause of death [1, 2]. In Italy, and other European countries, there are over 200.000 new cases of cerebrovascular disorders (CDs) annually, with the incidence of stroke in young adults peaking at a rate of ten cases per 100.000 inhabitants [3]. CDs in young adults are etiologically heterogeneous and the main clinical challenge in their management remains the identification of the causes. These may be the result of shared environmental and genetic factors, leaning toward the hereditary ones in young adults [4]. Single gene diseases that may present with stroke or transient ischemic attack (TIA), include Fabry’s disease (FD) [5], a rare X-linked inborn error of glycosphingolipids metabolism, caused by mutations in the alpha-galactosidase A (GLA) gene, resulting in the reduced production of the enzyme α-galactosidase A. In FD, the enzymatic deficiency leads to lysosomal accumulation of neutral glycosphingolipids in all tissues, particularly in vascular endothelial cells [6]. This is consistent with the natural history of FD that frequently includes the occurrence of different CDs, such as TIAs and stroke, even at very young age in both genders, and the appearance of white matter lesions (WMLs) on brain MRI due to cerebral vasculopathy [6–9]. Since patients with definite FD may be safely treated with a specific enzyme replacement therapy (ERT) with benefit, a timely diagnosis has therapeutic and prognostic implications [8, 10].
Recently, numerous studies investigating FD prevalence in particular populations at risk, such as patients with juvenile stroke, cryptogenic renal and/or cardiac disturbances, have been undertaken [11]. In particular, in the first screening for FD, in patients with juvenile stroke in Germany, a high number of FD patients (about 4 %) have been detected [12]. However, a subsequent Belgian study, conducted on a smaller sample of patients with stroke of unknown origin, was unable to identify any FD patient [13]. Considering these findings, we conducted the present, exploratory study to investigate the prevalence of FD in patients with juvenile stroke in northern Sardinia, a Mediterranean island, with up to now unrecognized FD. Judging precedent inclusion criteria too restrictive, with respect to the CDs qualifying events (QEs), we considered not only cryptogenic ischemic stroke but also the other classes of CDs included in the TOAST classification (Trials of ORG 10172 in Acute Stroke Syndromes) [14, 15] Moreover, we included patients with WMLs at brain MRI, possibly related to cerebral vasculopathy.
Methods
Design of the study
The Exploratory Fabry Cerebrovascular disease Screening is the first study designed for assessing the occurrence of FD in Sardinia. We prospectively analyzed patients with CDs admitted to our neurological ward, at Sassari University, in north Sardinia, which allows access to about 100 patients with CDs per year. Among these patients, approximately 20 % are people aged between 18 and 55 years of age. People admitted to our neurological ward come mainly from two north Sardinia areas, which in 2014 had about 480.000 inhabitants and 260.000 people aged between 18 and 55 years of age (2011 population census, ISTAT findings). Inclusion criteria were the following: age between 18 and 55 years of age at the time of the QE, and one of the following QEs, ischemic stroke (IS), TIA, intracerebral haemorrhage (ICH), WMLs at brain MRI possibly due to cerebral vasculopathy, and cerebral venous thrombosis (CVT). We considered the definition of stroke devised by the World Health Organization in the 1970s, “neurological deficit of cerebrovascular cause that persists beyond 24 h” [16]. Considering the multi-tissues involvement in FD patients, with regard to ischemic stroke we chose to consider all the other classes of CDs included in the TOAST classification [14, 15]. Moreover, we included patients with non-traumatic ICH, defined as every spontaneous bleeding into the brain [17]; patients with TIA, defined as an episode with stroke-like symptoms lasting no more than 24 h, [18] and patients with WMLs at brain MRI, even in absence of overt clinical manifestations when possibly due to cerebral vasculopathy [19–21]. CVT was defined as a thrombosis in one or more of cerebral veins.
A diagnosis of definite or uncertain FD was made on the basis of characteristic phenotypic or biochemical features of patients, according to recent diagnostic criteria for FD in adults [22, 23].
Our primary objective was to estimate the prevalence of positive DNA mutations or potentially pathogenic single nucleotide polymorphisms in the GLA gene in an unselected, consecutive group of patients with one or more of the chosen QEs. Our secondary objective was to assess, in each patient, the clinical presentation also in relation to others associated risk factors for CDs, and to point out the specific category of the QE, with special attention to patients with FD, in order to identify possible clues useful in clinical practice.
Participants and informed consent
All participants enrolled in the study provided written informed consent for enzymatic and genetic testing, and for the publication of individual clinical details. A detailed personal and familiar history, with particular attention to cerebrovascular risk factors and the occurrence of signs and symptoms indicative of FD were noticed. The study protocol was approved by the Human Ethics Review Committees of Sassari and Pavia (Prot. N° P-20070030590).
Biochemical studies and genetic analysis
A blood sample on Heparin and on EDTA was drawn from all patients, and sent to the Molecular Genetics Laboratory of the Institute of Recovery and Care of Scientific Character (IRCCS) Foundation “S. Matteo”, Pavia. Our patients were also integrated into the polycentric Italian study GIMAF (Interdisciplinary Group for Anderson Fabry Disease). Enzyme alpha-galactosidase A activity was assessed in plasma and leucocytes by spectrofluorimetry, considering normal α-galactosidase A level higher than 2 nmol/mL/h in plasma or 20 nmol/mg prot/h in leucocytes. DNA analysis was done for each of the seven exons of GLA gene, amplified by polymerase chain reaction (PCR). Each PCR product was analyzed by denaturing high-performance liquid chromatography (dHPLC), with a sensibility of 66–98 %, searching for heteroduplex and was examined amplicons with heteroduplex profile by automated DNA sequencing. Concentration of lyso-Gb3 in plasma was measured by tandem mass spectrometry in patients with positive DNA mutations or potentially pathogenic single nucleotide polymorphisms in the GLA gene, and in their relatives.
We used the centralized database of the GIMAF study and a database on our website, including data on clinical variables.
Results
We identified 187 eligible patients between April 2008 and April 2012. Nine patients were excluded: three patients because they did not give informed consent, the others 6 because they did not fulfill the requirements for definite cerebrovascular events. We included 178 patients (105 women, and 73 men). The average age at recruitment was 48.4+/−22.8 years of age, while the mean age at onset of CDs symptoms was 43.3 years of age (range 20–55 years). The onset of the QE for patients with WMLs refers to the first documentation of the WMLs at brain MRI. None of the patients had previous personal or familiar diagnosis of FD.
Two patients tested positive at the genetic GLA tests: one man aged 41, included for WMLs, who showed a known GLA mutation (R227Q), and severe cardiac and renal involvement, and a woman aged 52, included for recurrent, cryptogenic ischemic stroke, who carried the D313Y mutation of GLA gene and presented with combined cardiac and renal involvement. (Table 1) Moreover, the man had shown acroparesthesias since childhood together with frequent pain crisis, which mainly occurred while doing physical activity and during fever. Also, at brain MRI, he showed the pulvinar sign in T1 sequence, and dolichoectasia of vertebrobasilar artery [24]. The woman, instead, had a first occurrence of stroke at the age of 48, with the brain MRI showing multiple WMLs. Also, she had hypertrophic cardiomyopathy and renal damage with proteinuria and increased serum creatinine. In the man with the known GLA mutation (R227Q), alpha-galactosidase A activity was pathologically decreased to 0.5 μmol/l/h (reference, ≥ 2 μmol/l/h) and the concentration of lyso-Gb3 in plasma was increased to 33.3 ng/ml (reference, ≤1.8 ng/ml). In the woman carrying the D313Y variant, instead, alpha-galactosidase A activity was decreased in plasma, while the concentration of lyso-Gb3 was within normal limits.
Considering the distribution of QEs: 107 patients presented IS (59 women and 48 men), 30 TIA, 30 WMLs, eight ICH and three women CVT. Table 2 shows the etiopathogenetic TOAST classes we found. Patients with WMLs came to our attention because of different neurological disturbances: 11 of them showed gait disturbances, ten headache, six vertigo and dizziness, one an epileptic seizure, one an acute confusion state, one syncopal episodes.
With regard to cardiovascular risk factors, we found a high percentage of hypertension, (59.5 %) and hyperlipidemia (46.6 %), overweight (47.6 %), smoking (24.2 %) and hyperhomocysteinemia (14 %).
Of note, our FD patients were included in the study on average 5 years after the qualifying event. During this time frame, in these patients renal damage progressed with increase of proteinuria and serum creatinine
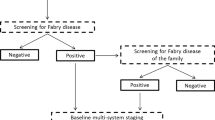
We also analyzed 22 relatives (12 women and ten men) of the two probands. In the first family group, out of 16 relatives examined, two women and three men resulted affected. In the second family, out of the six relatives studied, two resulted positive to FD at genetic tests, one man and one woman. The clinical features of the two families are shown in Table 3 and the family trees are presented in Fig. 1.

Family trees: Family tree (a) and family tree (b). Probands are indicated by arrows. Patients are numbered as in Table 3
Discussion
Several studies indicate that patients with FD have a 20-fold increased risk of ischemic stroke and TIA compared to general population [24–26]. Multiple CDs arising in young and middle-aged adults have been described in FD. Stroke is the most frequent, and can develop either as a consequence of cardiac embolism, or due to primary cerebral vasculopathy. In patients with FD, the cerebral vasculopathy is related to damage in small and large blood vessels, mainly at level of vertebro-basilar circulation [24, 26]. In a cohort study of 2446 patients from the Fabry Registry, 138 patients experienced stroke (about, 5.6 %). Half of these patients experienced their first stroke before being diagnosed with FD, and 70 % before any renal or cardiac disturbance [9]. Thus, it is important to search for FD in patients with early onset CDs. In literature, there is a wide variability, from 4 to 0 %, in the reported prevalence of FD in patients with cryptogenic stroke (Table 2) [12, 13, 27–32]. This is probably due to different methodological approaches, in particular the choice of different QEs, and the size of the sample analyzed. In our study, we chose to include several kinds of CDs, as previously detailed. In other studies, [33, 34] a wide range of different methods were used, in particular as regards the choice of QEs. The most recent study on prevalence of FD in young adults with CDs comes from the multinational European study Stroke in Young Fabry Patients (Sifap) [35]. This study, in a cohort of 5023 patients, aged between 18 and 55 years, found that the prevalence of definite FD was 0.5 %, with an adjunctive 0,4 % prevalence for probable FD, including in this definition patients carrying the D313Y variant [36]. In our study, according to recent diagnostic criteria in adults [22, 23], comprehensively, we found a 0,9 % prevalence of FD in young people with CDs, namely, 0,45 % of definite FD, plus 0,45 % of uncertain FD, including the patient with the D313Y variant genotype.
This is the first study that documents the occurrence of FD in neurologic patients in Sardinia island. Our result places in a middle position between the 4 % prevalence firstly reported in the study by Rolfs [12] (probably, overestimated), and the studies that found no FD patients in their clinical records (probably, underestimated) [13, 28]. In our study, two patients with a GLA gene variant were identified: a man, included for the occurrence of WMLs possibly due to cerebral vasculopathy at brain MRI, who showed the known GLA mutation (Arg227Gln), associated with a classical phenotype of FD. This patient had a profound decrease of alpha-galactosidase A activity in plasma and leukocytes, and an increased concentration of lyso-Gb3 in plasma; and a woman, included for recurrent ischemic stroke, who carried the D313Y GLA gene variant, commonly defined as polymorphism and reportedly associated with lower alpha-galactosidase A level in plasma and normal GLA enzyme activity in leukocytes, causing the so called “pseudodeficiency” [36]. In this woman, the concentration of lyso-Gb3 in plasma was within normal limits. Recently, this mutation, that was formerly reported as nonpathogenic, has been associated with multifocal WMLs at brain MRI and exclusive neurologic manifestations [37]. Of note, mono-organic manifestations have been frequently described with prominent cardiac or renal manifestations in FD [37]. Moreover, several studies reported an association of D313Y with other typical FD manifestations, such as stroke, renal failure, peripheral neuropathty, or hypertrophic cardiomyopathy [27, 33, 38]. In particular, a recent prospective study including 625 patients with cerebral ischemia, aged between 18 and 55 years, reported that this gene variant was associated with cryptogenic stroke [33]. Notably, the occurrence of a high proportion of FD in recurrent stroke is suggested by our findings and it has also been reported in the German and Belgian studies (Table 2) [12, 13, 34, 35].
Moreover, an analysis of studies in literature indicates that frequent etiologies of ischemic stroke in patients with FD, according to the TOAST classification, include the following: cryptogenic, cardioembolic, other determined and lacunar [12, 13, 27–35]. Interestingly, in literature, FD diagnosis has been never reported in patients with atherothrombotic strokes. FD diagnosis, instead, has also been reported in patients with TIA, ICH, occurrence of WMLs at brain MRI and CVT. We emphasize the potential clinical relevance of searching FD in patients with only WMLs. This is related to the continuing search for clinical or paraclinical markers useful for an early diagnosis in FD patients, and consequently a timely ERT in definite FD patients [10, 22, 23], with the possibility of delaying the natural course of the disease.
Sixteen per cent of our patients were included for WMLs at brain MRI, and we found our first patient in this group. WMLs are quite common in the elderly general population, but they are distinctly uncommon among the general population under 55 years of age [39]. A high prevalence of WMLs at MRI and silent infarcts were found in several studies in patients with cerebrovascular and cardiovascular disorders [39–41]. They have been found in patients under 49 years of age, in 8 % in the Framingham Offspring Study and the Helsinki Young Stroke Registry [41, 42]. Putaala et al. reported a 7 % prevalence of WMLs in a similar range of age [39]. Moreover, several studies reported a high prevalence of WMLs in FD patients, reaching 100 % after 55 years of age [42, 43]. Taken together, these findings indicate the need for a careful search for FD in this population. Further details on the distribution of FD diagnosis in relation to the inclusion criteria of our patients are reported in Table 2. Moreover, analyzing cardiovascular risk factors in our study population, we found a high percentage of hypertension, hyperlipidemia, overweight/obesity and hyperhomocysteinemia, in line with other similar screening studies.
Conclusions
Our data indicate the necessity to evaluate FD prevalence in patients with cerebrovascular disease in young age in the entire Sardinia island. In particular, the diagnosis of FD should be considered in patients with cryptogenic stroke and in patients with WMLs at brain MRI possibly related to cerebral vasculopathy, especially if other FD related symptoms are present. Attention should also be paid to recurrent stroke, which may hide a large proportion of FD patients. In patients at risk, a complete and detailed personal and family history should be obtained, together with an accurate search for clinical features indicative of FD at physical examination. Since FD is now a potentially treatable clinical condition, an early diagnosis may have relevant prognostic implications for patients and their relatives. Neurologists may have a prominent role in an early identification of this pathology.
Abbreviations
- CDs:
-
cerebrovascular dieases
- CKD:
-
chronic kidney disease
- CVT:
-
cerebral venous thrombosis
- ERT:
-
enzyme replacement therapy
- FD:
-
Fabry’s disease
- ICH:
-
intracerebral haemorrhage
- QEs:
-
qualifying events
- TIA:
-
transient ischemic attack
- WMLs:
-
white matter lesions
References
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and Stroke statistics-2011 update: a report from the American Heart association. Circulation. 2011;123:e18–e209.
Lackland DT, Roccella EJ, Deutsch AF, Fornage M, George MG, Howard G, et al. Factors influencing the decline in stroke mortality: a statement from the American Heart Association/American Stroke Association. Stroke. 2014;45:315–53.
Marini C, Totaro R, De Santis F, Ciancarelli I, Baldassarre M, Carolei A. Stroke in young adults in the community-Based L’Aquila registry: incidence and prognosis. Stroke. 2001;32:52–6.
Ferro JM, Massaro AR, Mas JL. Aetiological diagnosis of ischaemic stroke in young adults. Lancet Neurol. 2010;9:1085–96.
Meschia JF, Worrall BB, Rich SS. Genetic susceptibility to ischemic stroke. Nat Rev Neurol. 2011;7:369–78.
Feldt-Rasmussen U. Fabry Disease and Early Stroke. Stroke Res Treat. 2011;2011:615218.
Fellgiebel A, Müller MJ, Ginsberg L. CNS manifestations of Fabry’s disease. Lancet Neurol. 2006;5:791–5.
Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U, et al. Fabry disease: a review of current management strategies. QJM. 2010;103:641–59.
Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry registry. Stroke. 2009;40:788–94.
Biegstraaten M, Arngrimsson R, Barbey F, Boks L, Cecchi F, Deegan PB, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. 2015;10:36.
Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet. 2010;47:217–22.
Rolfs A, Bottcher T, Zschiesche M, Morris P, Winchester B, Bauer P, et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005;366:1794–6.
Brouns R, Sheorajpanday R, Braxel E, Eyskens F, Baker R, Hughes D, et al. Middelheim Fabry Study (MiFaS): a retrospective Belgian study on the prevalence of Fabry disease in young patients with cryptogenic stroke. Clin Neurol Neurosurg. 2007;109:479–84.
Adams Jr HP, Bendixen BH, Kappelle LJ, Biller J, Love BB. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993;24:35–41.
The Publications Committee for the Trial of ORG 10172 in Acute Stroke Treatment (TOAST) lnvestigators. Low Molecular Weight Heparinoid, QRG 10172 (Danaparoìd), and outcome After Acute Ischemic Stroke. JAMA. 1998;279:1265–72.
Kidwell CS, Warach S. Acute ischemic cerebrovascular syndrome: diagnostic criteria. Stroke. 2003;34:2995–8.
Morgenstern LB, Hemphill 3rd JC, Anderson C, Becker K, Broderick JP, Connolly Jr ES, et al. Guidelines for the Management of spontaneous intracerebral haemorrhage :a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2010;41:2108–29.
Siket MS, Edlow JA. Transient ischemic attack: reviewing the evolution of the definition, diagnosis, risk stratification, and management for the emergency physician. Emerg Med Clin North Am. 2012;30:745–70.
Chen Y, Wang A, Tang J, Wei D, Li P, Chen K, et al. Association of white matter integrity and cognitive functions in patients with subcortical silent lacunar infarcts. Stroke 2015; 46: 1123-6.
Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Arch Neurol. 1987;44:21–3.
Pantoni L, Inzitari D. New Clinical Relevance of Leukoaraiosis. Stroke. 1998;29:543.
Van der Tol L, Cassiman D, Houge G, Janssen MC, Lachmann RH, Linthorst GF, et al. Uncertain diagnosis of Fabry disease in patients with neuropathic pain, angiokeratoma or cornea verticillata: consensus on the approach to diagnosis and follow-up. JIMD Rep. 2014;17:83–90.
Smid BE, van der Tol L, Cecchi F, Elliott PM, Hughes DA, Linthorst GE, et al. Uncertain diagnosis of Fabry disease: consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int J Cardiol. 2014;177:400–8.
Fancellu L, Deiana GA, Sechi GP. Teaching neuroimages: Neuroimaging leads to recognition of previously undiagnosed Fabry disease. Neurology. 2010;75:e28.
Schiffmann R. Fabry disease. Pharm Ther. 2009;122:65–77.
Mitsias P, Levine SR. Cerebrovascular complications of Fabry’s disease. Ann Neurol. 1996;40:8–17.
Wozniak MA, Kittner SJ, Tuhrim S, Cole JW, Stern B, Dobbins M, et al. Frequency of unrecognized Fabry disease among young European-American and African-American men with first ischemic stroke. Stroke. 2010;41:78–81.
Sarikaya H, Yilmaz M, Michael N, Miserez AR, Steinmann B, Baumgartner RW. Zurich Fabry study – prevalence of Fabry disease in young patients with first cryptogenic ischaemic stroke or TIA. Eur J Neurol. 2012;19:1421–6.
Saposnik G, Lanthier S, Mamdani M, Thorpe KE, Melo M, Pope K, et al. Fabry’s disease: a prospective multicenter cohort study in young adults with cryptogenic stroke. Int J Stroke. 2012;7:265–73.
Dubuc V, Moore DF, Gioia LC, Saposnik G, Selchen D, Lanthier S. Prevalence of Fabry Disease in Young Patients with Cryptogenic Ischemic Stroke. J Stroke Cerebrovasc Dis. 2013;22:1288-92.
Marquardt L, Baker R, Segal H, Burgess AI, Poole D, Hughes DA, et al. Fabry disease in unselected patients with TIA or stroke: population-based study. Eur J Neurol. 2012;19:1427–32.
Shi Q, Chen J, Pongmoragot J, Lanthier S, Saposnik G. Prevalence of Fabry Disease in Stroke Patients-A Systematic Review and Meta-analysis. J Stroke Cerebrovasc Dis. 2014;23:985–92.
Baptista MV, Ferreira S, Pinho-E-Melo T, Carvalho M, Cruz VT, Carmona C, et al. Mutations of the GLA gene in young patients with stroke: the PORTYSTROKE study–screening genetic conditions in Portuguese young stroke patients. Stroke. 2010;41:431–6.
Brouns R, Thijs V, Eyskens F, Van den Broeck M, Belachew S, Van Broeckhoven C, et al. Belgian Fabry study:prevalence of Fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke. 2010;41:863–8.
Rolfs A, Fazekas F, Grittner U, Dichgans M, Martus P, Holzhausen M, et al. Acute cerebrovascular disease in the young: the Stroke in Young Fabry Patients study. Stroke. 2013;44:340–9.
Froissart R, Guffon N, Vanier MT, Desnik RJ, Maire I. Fabry disease: D313Y is an alpha galactosidase A sequence variant that cause pseudodeficient activity in plasma. Mol Genet Metab. 2003;80:307–14.
Lenders M, Duning T, Schelleckes M, Schmitz B, Stander S, Rolfs A, et al. Multifocal White matter lesions Associated with the D313Y mutation of the alfa-galactosidase A gene. PLoS One. 2013;8:e55565.
Gaspar P, Herrera J, Rodrigues D, Cerezo S, Delgado R, Andrade CF, et al. Frequency of Fabry disease in male and female haemodialysis patients in Spain. BMC Med Genet. 2010;11:19.
Putaala J, Kurkinen M, Tarvos V, Salonen O, Kaste M, Tatlisumak T. Silent brain infarcts and leukoaraiosis in young adults with first-ever ischemic stroke. Neurology. 2009;72:1823–9.
Vermeer SE, Longstreth Jr WT, Koudstaal PJ. Silent brain infarcts: a systematic review. Lancet Neurol. 2007;6:611–9.
Das RR, Seshadri S, Beiser AS, Kelly-Hayes M, Au R, Himali JJ, et al. Prevalence and correlates of silent cerebral infarcts in the Framingham offspring study. Stroke. 2008;39:2929–35.
Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, et al. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology. 1993;43:1683–9.
Fellgiebel A, Muller MJ, Mazanek M, Baron K, Beck M, Stoeter P. White matter lesion severity in male and female patients with Fabry disease. Neurology. 2005;65:600–2.
Acknowledgements
The authors thank all the patients, and Shire spa Italy for the financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
In the past five years Prof. GianPietro Sechi, received funding from Shire HGT for research purposes. The other authors declare that they have no competing interests.
Authors’ contributions
LF carried out the patient selection and collected informed consent and sample in the screening, participated in design and coordination of the study, and draft of the manuscript. WB participated in the design and helped to draft the manuscript. IR participated in the statistical analysis and helped to draft the manuscript. AP participated in the patient selection and statistical analysis. GAD carried out the patient selection and collected informed consent and sample in the screening. ES carried out the patient selection. DEP carried out the patient selection. ALR carried out the patient selection. RD participated in the patient selection and statistical analysis. AS carried out the immunoassays. MT participated in the sequence alignment. PC carried out the molecular genetic studies, participated in the sequence alignment. EA participated in its design and coordination, and helped to draft the manuscript. GPS participated in the design, coordination and draft of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fancellu, L., Borsini, W., Romani, I. et al. Exploratory screening for Fabry’s disease in young adults with cerebrovascular disorders in northern Sardinia. BMC Neurol 15, 256 (2015). https://doi.org/10.1186/s12883-015-0513-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-015-0513-z