
Abstract
Background
Immune Checkpoint Inhibitors (ICPIs) are promising new drugs in treatment of advanced tumours targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death protein-1 (PD1) or its ligand (PDL-1). Ipilimumab is a monoclonal antibody targeting the CTLA-4 receptor used in treatment of metastatic melanoma. By increasing activity of the immune system, ICPIs lead to immune-related adverse events, such as dermatitis, colitis or hepatitis. ICPIs-related kidney adverse events are rare and acute tubulointerstitial nephritis with or without granuloma have mainly been reported.
Case presentation
We report a case of acute kidney injury in a patient with melanoma treated by ipilimumab. Kidney biopsy revealed acute interlobular and juxtaglomerular granulomatous arteritis, which has not yet been reported in patients treated by ICPIs. Kidney function partially recovered after ipilimumab discontinuation and oral prednisone. Unfortunately, the patient died a few months later from progression of his melanoma.
Conclusion
This case highlights a new mechanism of acute kidney injury related to ICPIs and supports the interest of kidney biopsy in case of ICPIs related acute renal failure.
Similar content being viewed by others

Background
Immune check point inhibitors (ICPIs) are monoclonal antibodies stimulating the immune system by blocking the coinhibitory receptors on T cells, leading to antitumoral response. This class of drugs includes cytotoxic T-lymphocyte antigen 4 (CTLA-4) inhibitors, such as ipilimumab, programmed cell death protein 1 (PD1) inhibitors and programmed cell death protein-ligand 1 (PDL-1) inhibitors. These molecules disrupt immune tolerance, leading to anti-tumor immunity but also inflammatory side effects, named immune-related adverse events (IRAEs), such as dermatitis, colitis, hepatitis or endocrinopathies [1].
ICPI-related acute kidney injuries are reported in 0 to 4% of treated patients [2]. The main pathologic lesion is interstitial nephritis, with or without granuloma [2, 3]. A few cases of immune glomerulonephritis have also been described [4, 5].
Here, we describe the first case of ipilimumab-induced renal granulomatous arteritis in a patient with melanoma.
Case presentation
A 70-year-old man with anal metastatic melanoma was admitted on December 2017 for severe asthenia.
He was treated on August 2016 by abdominoperineal resection and bladder reconstruction. Serum creatinine was 1.0 mg/dl before the surgery. He presented an acute kidney injury in a context of urinary tract infection one month later (serum creatinine 2.9 mg/dl). Kidney function recovered partially (serum creatinine 1.4 mg/dl - Fig. 1). In September 2016, he received nivolumab, a monoclonal anti-PD-1 receptor antibody. In October 2017, progression of the disease was detected on a pelvic MRI and treatment was switched by ipilimumab, a monoclonal anti-CTLA-4 receptor antibody, 3 mg/kg every three weeks. Serum creatinine was then at 1.7 mg/dl. He had no other medical history nor took any other drugs.

Evolution of kidney function
Approximately ten days after completion of his third cycle, he presented with a history of one-week fatigue. At admission, blood pressure was 116/60 mmHg and physical examination revealed no particularity. Laboratory findings revealed an acute renal failure with serum creatinine rising to 5.8 mg/dl (Fig. 1), a 24-h urine total protein excretion of 1.0 g and an inflammatory syndrome. Urine cytology showed leukocytes (100/mm3) and red blood cells (50/mm3). Ultrasonography excluded urinary tract obstruction. All laboratory results are detailed in Table 1.
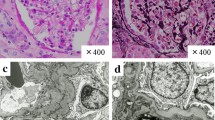
A percutaneous kidney biopsy was performed and revealed on light microscopy interlobular and juxtaglomerular noncaseating granulomatous arteritis (Fig. 2), accompanied by severe interstitial inflammation. The glomeruli appeared unremarkable. Immunohistochemistry for IgG, IgA, IgM, C3, C4, kappa and lambda chains was negative. Stainings for fungi and acid-fast bacilli were also negative. Acute segmental and focal granulomatous arteritis induced by ipilimumab was diagnosed.

Kidney biopsy showing destruction of arteries by epithelioid cell granulomas (Masson’s trichrome (a) and H&E staining (b)). (a-b) Light microscopy. (a) Juxtaglomerular (↘) and periarteriolar (*) non caseating granulomas. Glomeruli were unremarkable (Masson trichrome staining; original magnification, × 200). (b) Segmental necrotizing granulomatous arteritis (H&E staining; original magnification, × 400)
Antinuclear antibodies and anti-neutrophil cytoplasmic antibodies (ANCA) were negative (anti-MPO < 3.2 and anti-PR3 < 2.3). Angiotensin convertase enzyme (45 UI/L), calcemia (2.24 mmol/L) and chest CT-scan were normal. These negative exams and the absence of cutaneous or peripheral lymph node involvement allowed us to exclude sarcoidosis.
Ipilimumab was immediately discontinued, and the patient was treated with oral prednisone at a dose of 1 mg/kg/day for one month, followed by gradual discontinuation over 4 weeks. Serum creatinine rapidly improved to 2.8 mg/dl two weeks after steroid introduction. One month after steroid discontinuation, serum creatinine remained stable (2.8 mg/dl - Fig. 1). Unfortunately, the patient died a few months later from progression of his melanoma.
Discussion and conclusions
We report here the first case of kidney vasculitis induced by ICPIs. According to the Chapel Hill International Consensus Conference (CHCC) nomenclature, vasculitides are classified based on vessels size into large (aorta and its main branches), medium (coronary, mesenteric, intra-renal), and small (arterioles and capillaries) vessel disease [6]. Renal small-vessel vasculitides, with or without granuloma, have been described in Giant Cell Arteritis (GCA), polyarteritis nodosa [7], sarcoidosis [8] or ANCA-associated vasculitis. Renal acute necrotizing arteritis with granulomatous perivascular infiltrate has also been reported in a patient with metastatic melanoma treated with encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) [9].
ICPIs-related vasculitis, predominantly large-vessel vasculitis and vasculitis of the nervous system, have already been described. Daxini et al. reported 20 cases of vasculitis associated with ICPIs, involving small (digital or retinal vasculitis, granulomatosis with polyangiitis), medium (nervous system) or large vessels (giant cell arteritis), but no case of renal granulomatous arteritis [10]. Goldstein et al. reported two cases of polymyalgia rheumatica and giant cell arteritis (PMR/GCA) following a treatment by ipilimumab [11]. These cases suggest that lymphocyte T costimulation blockers, such as abatacept, may have beneficial therapeutic implications in the management of PMR/GCA.
Cortazar et al. reported kidney pathology in 13 patients with ICPIs-induced acute kidney injury (AKI). Kidney biopsies revealed acute interstitial nephritis (AIN) in 12 patients, with granulomatous features in 3 patients, whereas the last patient presented acute thrombotic microangiopathy (TMA) without AIN [3]. Many cases of AIN have been reported [12, 13], while immune-related glomerulonephritis [4] or nephrotic syndrome [5] have occasionally been described. To our knowledge, we report here the first case of renal granulomatous arteritis induced by ipilimumab.
PD-1 and CTLA-4 are known as immune checkpoints. They prevent activation of T-lymphocyte by binding their respective receptors on T-cells. Cancer cells overexpress PD-1 and/or CTLA-4 proteins to down-regulate T-cells and allow tumor growth and metastasis. Checkpoint inhibitors are monoclonal antibodies (anti-PD-1 and anti-CTLA-4) that are used as an immunotherapy to destroy cancer cells by allowing T-cells to proliferate within the tumor microenvironment. Two mechanisms are proposed to explain the pathophysiology of AIN in patients exposed to ICPIs. First, ICPIs may disrupt tolerance to exogenous drug antigens, allowing reactivation of memory T-cells previously primed by exogenous drug exposure. Second, as the CTLA-4 and PD-1 pathways are normally employed to limit unwanted autoimmunity, interference with CTLA-4 and PD-1 pathways may disrupt peripheral tolerance of self-antigens by blunting activation of self-reactive T-cells. These two pathways may lead to migration of effector T-cells into the kidney and development of acute tubulointerstitial nephritis [14]. Our patient did not take any other drug. So, the second pathway can presumably be involved in vascular T-cells influx leading to development of granulomas in our patient.
Interestingly, single nuclear polymorphisms of CTLA4 have been reported to be associated with ANCA-positive small vessels vasculitis (AAV) [15]. These genetic polymorphisms may lead to hyperreactivity of T cells and thus contribute to the pathogenesis of AAV [16]. Even though ANCA were negative, we may suppose that pathophysiology of renal granulomatous arteritis in our patient was directly linked to CTLA-4 blocking.
Management of ICPIs-induced AKI is still challenging. The efficacy of glucocorticoids in the treatment of AIN remains controversial in the absence of randomized trials. However, based on case reports and case series, Izzedine et al. suggest that management of immune-related renal toxicities should consist in prompt treatment discontinuation and steroid therapy [2]. In their study, Cortazar et al. reported 2 total remissions (serum creatinine < 0.35 mg/dl above baseline value) and 7 partial remissions (serum creatinine > 0.35 mg/dl above baseline value) among 10 patients receiving a steroid treatment [3]. In our patient, ipilimumab discontinuation and steroid therapy led to partial remission, with nevertheless tumor progression leading to death in the absence of effective substitution treatment for advanced melanoma. This highlights another important consideration, which is the possibility to continue or reintroduce ICPI therapy in patients with kidney injury. In their series, Cortazar et al. reported one patient with relatively stable renal function despite continuation of ipilimumab without steroid therapy [3]. Izzedine et al. suggest the possibility to continue ICPI in case of a grade 1 renal event and to postpone the treatment until plasma creatinine decreases to at least grade 1, in case of grade 2–3 renal event [2].
Finally, whether patients with IRAEs have a better anti-tumor response to ICPI therapy remains unknown. A recent study suggests that IRAEs may be associated with improved progression-free survival (PFS) in patients receiving anti-PD-1/PD-L1 therapy. Furthermore, in this study, the use of systemic corticosteroids does not appear to alter PFS [17].
In conclusion, we report here the first case of ipilimumab-induced renal granulomatous arteritis, a new entity of ICPIs’ immune related adverse events. This case highlights the importance of rapid kidney biopsy to elucidate the cause of kidney injury related to ICPIs and promptly start steroid therapy.
Availability of data and materials
Further clinical data and images to support this case are available from the corresponding author upon reasonable request.
Abbreviations
- AAV:
-
ANCA-positive small vessels vasculitis
- AIN:
-
Acute Interstitial Nephritis;
- AKI:
-
Acute Kidney Injury
- ANCA:
-
Anti-Neutrophil Cytoplasmic Antibodies
- CTLA-4:
-
Cytotoxic T-Lymphocyte Antigen-4
- GCA:
-
Giant Cell Arteritis
- ICPIs:
-
Immune Checkpoint Inhibitors
- IRAE:
-
Immune-Related Adverse Events
- PD1:
-
Programmed cell Death protein-1
- PDL-1:
-
Programmed cell Death protein-Ligand 1
- PMR/GCA:
-
Polymyalgia Rheumatica and Giant Cell Arteritis
- TMA:
-
Thrombotic Microangiopathy
References
Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with Cancer: a systematic review of case reports. Nishikawa H, éditeur. PLOS ONE 29 juill 2016;11(7):e0160221.
Izzedine H, Mateus C, Boutros C, Robert C, Rouvier P, Amoura Z, et al. Renal effects of immune checkpoint inhibitors. Nephrol Dial Transplant 26 déc. 2016; gfw382.
Cortazar FB, Marrone KA, Troxell ML, Ralto KM, Hoenig MP, Brahmer JR, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int sept. 2016;90(3):638–47.
Fadel F, Karoui KE, Knebelmann B. Anti-CTLA4 antibody–induced lupus nephritis. N Engl J Med 9 juill. 2009;361(2):211–2.
Kitchlu A, Fingrut W, Avila-Casado C, Chan CT, Crump M, Hogg D, et al. Nephrotic syndrome with Cancer immunotherapies: a report of 2 cases. Am J Kidney Dis oct. 2017;70(4):581–5.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international Chapel Hill consensus conference nomenclature of Vasculitides. Arthritis Rheum janv. 2013;65:1):1–11.
Maritati F, Iannuzzella F, Pavia MP, Pasquali S, Vaglio A. Kidney involvement in medium- and large-vessel vasculitis. J Nephrol août. 2016;29(4):495–505.
Agrawal V, Crisi GM, D’Agati VD, Freda BJ. Renal sarcoidosis presenting as acute kidney injury with granulomatous interstitial nephritis and Vasculitis. Am J Kidney Dis 1 févr. 2012;59(2):303–8.
Maanaoui M, Saint-Jacques C, Gnemmi V, Frimat M, Lionet A, Hazzan M, et al. Glomerulonephritis and granulomatous vasculitis in kidney as a complication of the use of BRAF and MEK inhibitors in the treatment of metastatic melanoma. Medicine (Baltimore) [Internet]. 23 juin 2017 [cité 1 nov 2018];96(25). Disponible sur: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5484213/
Daxini A, Cronin K, Sreih AG. Vasculitis associated with immune checkpoint inhibitors—a systematic review. Clin Rheumatol sept. 2018;37(9):2579–84.
Goldstein BL, Gedmintas L, Todd DJ. Drug-associated polymyalgia Rheumatica/Giant cell arteritis occurring in two patients after treatment with Ipilimumab, an antagonist of CTLA-4: concise communication. Arthritis Rheumatol mars. 2014;66(3):768–9.
Shirali AC, Perazella MA, Gettinger S. Association of Acute Interstitial Nephritis with Programmed Cell Death 1 inhibitor therapy in lung Cancer patients. Am J Kidney Dis août. 2016;68(2):287–91.
Izzedine H, Gueutin V, Gharbi C, Mateus C, Robert C, Routier E, et al. Kidney injuries related to ipilimumab. Invest New Drugs août. 2014;32(4):769–73.
Perazella MA. Checkmate: kidney injury associated with targeted cancer immunotherapy. Kidney Int. sept. 2016;90(3):474–6.
Kamesh L, Heward JM, Williams JM, Gough SCL, Chavele K-M, Salama A, et al. CT60 and +49 polymorphisms of CTLA 4 are associated with ANCA-positive small vessel vasculitis. Rheumatology 1 déc. 2009;48(12):1502–5.
Slot MC, Sokolowska MG, Savelkouls KG, Janssen RGJH, Damoiseaux JGMC, Cohen Tervaert JW. Immunoregulatory gene polymorphisms are associated with ANCA-related vasculitis. Clin Immunol juill. 2008;128(1):39–45.
Shafqat H, Gourdin T, Sion A. Immune-related adverse events are linked with improved progression-free survival in patients receiving anti-PD-1/PD-L1 therapy. Semin Oncol [Internet]. oct 2018 [cité 12 nov 2018]; Disponible sur: https://linkinghub.elsevier.com/retrieve/pii/S0093775418300095
Acknowledgements
Not applicable.
Funding
This study was not supported financially by any public, private or non-for-profit organization. No funding body support the design of the study and collection, analysis, interpretation of data, or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
ML, BD, AC and AF made substantial contributions to acquisition of data. ML, DB and DG made substantial contributions to conception and design. MH, CL, VH, AF and SG made substantial contributions to analysis and interpretation of data. ML, BD, and AC were involved in drafting the manuscript. DB, DG, MH, CL, VH, AF and SG were involved in revising it critically for important intellectual content. AF performed the histological examination of the kidney. All the authors gave final approval of the version to be published. All the authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient’s son for publication of this case report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lemoine, M., Dilly, B., Curie, A. et al. Ipilimumab-induced renal granulomatous arteritis: a case report. BMC Nephrol 20, 366 (2019). https://doi.org/10.1186/s12882-019-1552-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-019-1552-2