
Abstract
Background
The revised 2016 WHO-Classification of CNS-tumours now integrates molecular information of glial brain tumours for accurate diagnosis as well as for the development of targeted therapies. In this prospective study, our aim is to investigate the predictive value of MR-spectroscopy in order to establish a solid preoperative molecular stratification algorithm of these tumours. We will process a 1H MR-spectroscopy sequence within a radiomics analytics pipeline.
Methods
Patients treated at our institution with WHO-Grade II, III and IV gliomas will receive preoperative anatomical (T2- and T1-weighted imaging with and without contrast enhancement) and proton MR spectroscopy (MRS) by using chemical shift imaging (MRS) (5 × 5 × 15 mm3 voxel size). Tumour regions will be segmented and co-registered to corresponding spectroscopic voxels. Raw signals will be processed by a deep-learning approach for identifying patterns in metabolic data that provides information with respect to the histological diagnosis as well patient characteristics obtained and genomic data such as target sequencing and transcriptional data.
Discussion
By imaging the metabolic profile of a glioma using a customized chemical shift 1H MR spectroscopy sequence and by processing the metabolic profiles with a machine learning tool we intend to non-invasively uncover the genetic signature of gliomas. This work-up will support surgical and oncological decisions to improve personalized tumour treatment.
Trial registration
This study was initially registered under another name and was later retrospectively registered under the current name at the German Clinical Trials Register (DRKS) under DRKS00019855.
Similar content being viewed by others


Background
Gliomas are a very heterogenic group of tumours arising from glial cells in the central nervous system that are yet to be completely understood. Recent research advances have led to the characterization of different tumour subgroups based on molecular properties and have shown the necessity to individualize patient treatment according to each tumour entity. As the revised classification of brain tumours by the World Health Organization of 2016 gave an essential role to the specific genetic alterations, not only for prognosis or probability of chemosensitivity, but also for the actual classification of tumours, determining the molecular alterations of these tumours in a reliable way is the most important task in neuro-oncology today [1]. However, not all of the necessary molecular diagnostic methods are available throughout the world, and a medical expert in neuropathology may also be scarce. Hence the growing interest in developing tools to aid in the diagnosis and classification of brain tumours in a non-invasive and more efficient way. MR spectroscopy has long been a neuroradiological tool used to determine the metabolic alterations in different central nervous system pathologies [2,3,4]. Nonetheless, advances in MR imaging as well as in nuclear medicine imaging have lifted MR spectroscopy in the routine diagnostic workup of patients suggested to have a brain disease to another level. In many institutions, MR spectroscopy is used for research purposes only, or is regarded as a somewhat outed method with little diagnostic significance in addition to MRI [5,6,7]. Nonetheless, MR spectroscopy can deliver information beyond the metabolic profile of a lesion [5, 8].
Since 1990 several studies have been conducted to show the role of MR spectroscopy in the differentiation between various central nervous system lesions. In gliomas, these studies have been mostly based on the information provided by alterations in the metabolite ratios of i.e. Cho/Cr, Cho/NAA, and NAA/Cr [9,10,11,12,13]. However, the necessity of uncovering more information about the genetic profile of gliomas in a preoperative setting is the goal of research conducted in the field of MR spectroscopy [14, 15]
Following the discovery of IDH-mutations in humans, Choi et al. published a study in 2012 regarding the detection of 2-hydroxyglutarate specifically through MR spectroscopy in patients with gliomas, which accurately correlated with the presence of an IDH-mutation and the diagnosis of WHO-Grade II and III gliomas in adults [14]. This advance indicated that there is much more information hiding behind chemical shift imaging [15]. Nevertheless, MRS still has disadvantages which complicate its implementation into the routine workup of tumour patients. Some of these are the lack of experience in interpreting results, the need of a homogeneous magnetic field in order to discern the resonance frequencies of each specific metabolite, the low concentration of metabolites in tumour lesions in respect to water (e.g. by edema), making a weak signal given by the resonant frequencies, the requirement of the lesion not being in the proximity of bone structures (i.e. skull base) and the fact that more interest has been put into developing molecular diagnostics processes of tumour tissue itself [15,16,17].
Previous work done by our authors has shown that gliomas can be classified through MR spectroscopy characteristics [18], and has shown furthermore a feasible way in which a genetic profiling based on MR spectroscopy can be done with metabolic alterations identified by MR spectroscopy being correlated and classified according to certain aberrations in oncogenic pathways [19,20,21].
Objectives
In the present study, the main objective of this research study is to correlate spectroscopic and genetic signatures of gliomas, and to apply a machine learning algorithm through which an integrative automated and accurate diagnostic work-up of these tumours can be developed.
Methods/design
Study setting
The present study will be conducted at the Departments of Neurosurgery and Neuroradiology of the Medical Centre – University Freiburg. Our hospital is a tertiary care centre with referrals of a wide region that encompasses the borders of southwest Germany, northwest Switzerland and northeast France.
Trial design
The study is a 5-year, monocentre, single arm, prospective diagnostic study trial of patients undergoing diagnostics and treatment for WHO-Grade II, III and IV gliomas.
Trial status
The first patient was recruited on the 1st of August of 2016. The expected date of completion is May 2021.
Participants characteristics
Eligibility criteria
Patients undergoing diagnostics and treatment for WHO-Grade II, III and IV gliomas who match the following criteria will be included in the study: (1) age older than 18 years, (2) suspected glial tumour and (3) location of the lesion not near the skull base.
MRI and MRS will be performed by a team of radiology technician and neuroradiologist specially trained for the planning and analysis of MRI and MRS in brain tumours in the Department of Neuroradiology. Glioma surgery will be performed by the surgeons at the Department of Neurosurgery.
Interventions
Intervention description
Following written consent, patients with a suspected glioma will receive preoperative anatomical (T1-weighted sequences with and without contrast enhancement, T2- and T2-FLAIR-weighted sequences) and chemical shift imaging (CSI).
After imaging acquisition, patients will undergo surgical glioma resection. Intraoperative tissue samples of the contrast enhancing tumour part will be obtained for histopathological and genetic analysis.
Afterwards, spectral data will be matched to the final histopathologic diagnosis including metabolic profile as well as transcriptome analysis. The combined data of two thirds of the cohort will be used for training a machine learning approach, specifically an artificial neuronal network. The data of the remaining one third of the cohort will be consecutively used for validation of the algorithm (Fig. 1).

Concept of study design
Imaging acquisition
Patients will be undergoing preoperative anatomical and in-vivo chemical shift imaging at a 3 T MR Magnetom Prisma scanner (Siemens, Erlangen, Germany) in the Department of Neuroradiology, Medical Centre—University Freiburg (details in Fig. 1.) Chemical shift imaging will be performed using the manufacturer’s provided chemical shift imaging sLASER sequence with a TE = 40 ms (Table 1). Partitions with a volume of interest (VOI) of 5 × 5x15 mm3 are intended to allow a more precise assignment of the area for intraoperative tissue sampling planning.
A timely evaluation will be performed using Syngovia® software (Siemens, Erlangen, Germany) and metabolic maps of twelve metabolites will be calculated as ratio to the contralateral normal appearing matter (NAM) and overlaid on high resolution T2- weighted images (0.63 × 0.63 × 2 mm3). An additional analysis by LC-Model will be performed and compared to Syngovia® software, in order to analyse the reliability of Syngovia® software. This step will ensure that the manufacturer’s software is able to analyse the spectroscopic data without the necessity for special spectroscopic software. Neuroradiologists with more than 20 years’ experience oversaw the imaging acquisition.
Individual spectra will be baseline corrected and fitted for twelve metabolites. Spectral data will then be aligned to the anatomical imaging and manually segmented into the following regions: normal appearing matter (NAM), ventricles (V) and lesion (L). Voxels with less than 70% overlay to a specific region will be excluded from further analysis.
Tissue sampling
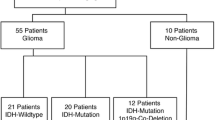
Navigation-based tissue sampling (Cranial Map Neuronavigation Cart 2, Stryker®, Freiburg, Germany) will be performed after previous planning by the surgical team at the Department of Neurosurgery of the Medical Centre—University Freiburg. Sampling locations will be aligned to the MR spectroscopy voxels by integration into the neuronavigation system. In high-grade glioma, sampling will be taken from the contrast-enhancing regions and in low-grade lesions from the T2-hyperintense regions. Tissue samples will immediately be snap-frozen in liquid nitrogen and processed. RNA will be prepared using the RNeasy Microarray Tissue kit (Qiagen®). An amount of 1.5 µg of RNA will be obtained for expression analysis. Neuropathological evaluation will be performed in the Department of Neuropathology, Medical Centre—University Freiburg according to their standards, including genome analysis (1p19q co-deletion and exome sequencing of IDH1/2).
Transcriptome analysis (RNA sequencing)
RNA integrity will be measured using the Agilent RNA Nano Assay Agilent Bioanalyser 2100 (http://www.home.agilent.com) according to the manufacturer’s instructions. Sequencing libraries will be prepared according to the manufacturer’s instructions and sequenced on an Illumina HiSeq4000 sequencer. The resulting 150 bp paired-end reads will be quality-checked with FastQC. Sequencing data will be mapped to the human genome (hg38, UCSC, https://genome.ucsc.edu) with STAR (https://github.com/alexdobin/STAR). Followed by the alignment, transcripts with less than 10 reads will be removed. The given RNA counts will be normalized by DESeq2 based on a negative binomial distribution model [22]. STAR-Fusion will further process the output generated by the STAR aligner (above described) to map junction reads and spanning reads to a reference annotation set (https://github.com/STAR-Fusion/).
Participant timeline
A participant timeline is shown in Fig. 2.

Participant timeline
Patient follow-up will be conducted according to our institution’s guidelines.
Sample size
The number of prospective cases to be analysed is n = 300. The prospective case number is based on the counselling by Dr. Harald Binder of the Institute of Medical Biometry and Statistics, Faculty of Medicine and Medical Centre—University of Freiburg.
The imaging dataset will be trained by genetic profiling and result in a new “radiogenomic classification” (n = 200 in training set). This new classifier will be compared to the histopathological “gold standard” and tested in an independent validation set (n = 100). Additional survival analysis will be performed by Cox Proportional-Hazards Model. We expect to enrol 300 patients in the study.
Data collection and management
Data management
All data material will be recorded with our institution’s own patient ID and will be unidentifiable outside the institution. The data will be stored electronically within our institution’s server and deleted 10 years after the project’s end.
Confidentiality
Only the primary investigators will have access to the patients’ entire information (names, birthdays and so forth). Data will be protected according to German and European laws. Data quality will be checked by two independent researchers of the neurosurgical team.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial or future use
All patients’ tumour samples (used for histopathological, genetic and molecular analysis) will be stored at the Tumour Bank of the Department of Neurosurgery, Medical Centre – University Freiburg according to our institution’s guidelines and adhering to German and European Laws.
Statistical methods
Biostatistics
The sample size has been previously determined by the Institute of Medical Biometry and Statistics based on prior experience [23]. It is of note, that there are to date no formal approaches for sample size calculation for complex multi-marker setting, as considered here.
The transcriptome data will be used to predict the expression subgroup of each patient. Data analysis will be performed by the Microenvironment and Immunology Research Laboratory at the Medical-Centre University of Freiburg. Obtained transcriptomic profiles will be analysed by the RawToViz pipeline (www.github.com/heilandd), and results will be available for the Vis_Labv.1.5 software (https://heilandd.shinyapps.io/Vis_Lab1/). To predict the subgroup affiliation, we use the state analysis based on a model first presented by Neftel et al in 2019 [24].
Artificial neural network algorithm
For training the neural network algorithm, 120 spectroscopic samples with 256 voxels (16 × 16 matrix) providing twelve metabolites each (input layer array of 120 × 256x12), resulting in 368,640 subsets, will be used. A deep learning approach will be used for uncovering metabolic patterns and linking these to a categorical output. In the first unsupervised step, spectroscopic data of the first half of the 120 samples will be analysed according to their correlation structure. Specifically, we will use partitioned deep Boltzmann machines (DBMs) [23], which are particularly suited for applications with small sample sizes, to learn the distribution of metabolite measurements in samples of different classes. This will result in one DBM per class, where each DBM provides a compression of the information into a small number of top-level hidden network nodes. Subsequently, measurements from a new patient could be fed into each DBM, resulting in several compressed representations. We intend to use an additional DBM for linking the joint output of the per-class DBMs to the class labels.
Interim analyses
An interim analysis is planned after n = 120 patients (30,720 voxels). The data of this cohort will then be used for the training of the neuronal network algorithm.
Plans to give access to the full protocol, participant level-data and statistical code
The research team intends to make the complete genomic and imaging datasets available for the whole research community by providing it on a suitable portal (i.e. The Cancer Genome Atlas (TCGA) as well as The Cancer Imaging Archive (TCIA)).
Discussion
Our study aims to develop a machine learning tool that can bring forth and correlate the metabolic signature of a glioma to its genetic profile. The metabolic signature will be derived from a readily available customized 3 T 1H chemical shift MR spectroscopy sequence. By using an open-software tool and a customized MRI sequence we intend to standardize the diagnostic work-up of gliomas and make it applicable even in places where genetic testing and therefore thorough diagnostic might be difficult to implement.
Availability of data and materials
The datasets and algorithms resulting and utilized in this study can be available upon request to the corresponding author.
Abbreviations
- MRS:
-
MR-spectroscopy: magnetic resonance spectroscopy, chemical shift imaging
- WHO:
-
World Health Organisation
- Cho:
-
Choline
- Cr:
-
Creatine
- NAA:
-
N-acetyl-aspartate
- IDH:
-
Isocitrate dehydrogenase
- CNN:
-
Convolutional neural network
- NAM:
-
Normal appearing matter
- VOI:
-
Volume of interest
- IDH:
-
Isocitrate dehydrogenase
- TE:
-
Echo time
- GS:
-
Gene significance
- MM:
-
Module membership
- kME:
-
Intramodule connectivity
- DBM:
-
Deep Boltzman machine
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20. https://doi.org/10.1007/s00401-016-1545-1.
Oz G, Alger JR, Barker PB, Bartha R, Bizzi A, Boesch C, et al. Clinical proton MR spectroscopy in central nervous system disorders. Radiology. 2014;270:658–79. https://doi.org/10.1148/radiol.13130531.
Tognarelli JM, Dawood M, Shariff MIF, Grover VPB, Crossey MME, Cox IJ, et al. Magnetic resonance spectroscopy: principles and techniques: lessons for clinicians. J Clin Exp Hepatol. 2015;5:320–8. https://doi.org/10.1016/j.jceh.2015.10.006.
Berrington A, Schreck KC, Barron BJ, Blair L, Lin DDM, Hartman AL, et al. Cerebral ketones detected by 3T MR spectroscopy in patients with high-grade glioma on an atkins-based diet. AJNR Am J Neuroradiol. 2019;40:1908–15. https://doi.org/10.3174/ajnr.A6287.
Lin A, Ross BD, Harris K, Wong W. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx. 2005;2:197–214. https://doi.org/10.1602/neurorx.2.2.197.
Hellström J, Romanos Zapata R, Libard S, Wikström J, Ortiz-Nieto F, Alafuzoff I, et al. The value of magnetic resonance spectroscopy as a supplement to MRI of the brain in a clinical setting. PLoS ONE. 2018;13:e0207336. https://doi.org/10.1371/journal.pone.0207336.
Verma A, Kumar I, Verma N, Aggarwal P, Ojha R. Magnetic resonance spectroscopy - revisiting the biochemical and molecular milieu of brain tumors. BBA Clin. 2016;5:170–8. https://doi.org/10.1016/j.bbacli.2016.04.002.
Bulik M, Jancalek R, Vanicek J, Skoch A, Mechl M. Potential of MR spectroscopy for assessment of glioma grading. Clin Neurol Neurosurg. 2013;115:146–53. https://doi.org/10.1016/j.clineuro.2012.11.002.
Bertholdo D, Watcharakorn A, Castillo M. Brain proton magnetic resonance spectroscopy: introduction and overview. Neuroimaging Clin N Am. 2013;23:359–80. https://doi.org/10.1016/j.nic.2012.10.002.
Majós C, Aguilera C, Alonso J, Julià-Sapé M, Castañer S, Sánchez JJ, et al. Proton MR spectroscopy improves discrimination between tumor and pseudotumoral lesion in solid brain masses. AJNR Am J Neuroradiol. 2009;30:544–51. https://doi.org/10.3174/ajnr.A1392.
Horská A, Barker PB. Imaging of brain tumors: MR spectroscopy and metabolic imaging. Neuroimaging Clin N Am. 2010;20:293–310. https://doi.org/10.1016/j.nic.2010.04.003.
Mader I, Roser W, Hagberg G, Schneider M, Sauter R, Seelig J, et al. Proton chemical shift imaging, metabolic maps, and single voxel spectroscopy of glial brain tumors. MAGMA. 1996;4:139–50.
Durmo F, Rydelius A, Cuellar Baena S, Askaner K, Lätt J, Bengzon J, et al. Multivoxel 1H-MR spectroscopy biometrics for preoprerative differentiation between brain tumors. Tomography. 2018;4:172–81. https://doi.org/10.18383/j.tom.2018.00051.
Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med. 2012;18:624–9. https://doi.org/10.1038/nm.2682.
Di Ieva A, Magnussen JS, McIntosh J, Mulcahy MJ, Pardey M, Choi C. Magnetic resonance spectroscopic assessment of isocitrate dehydrogenase status in gliomas: the new frontiers of spectrobiopsy in neurodiagnostics. World Neurosurg. 2020;133:e421–7. https://doi.org/10.1016/j.wneu.2019.09.040.
Kauppinen RA, Peet AC. Using magnetic resonance imaging and spectroscopy in cancer diagnostics and monitoring: preclinical and clinical approaches. Cancer Biol Ther. 2011;12:665–79. https://doi.org/10.4161/cbt.12.8.18137.
Cianfoni A, Law M, Re TJ, Dubowitz DJ, Rumboldt Z, Imbesi SG. Clinical pitfalls related to short and long echo times in cerebral MR spectroscopy. J Neuroradiol. 2011;38:69–75. https://doi.org/10.1016/j.neurad.2010.10.001.
Hagberg G, Burlina AP, Mader I, Roser W, Radue EW, Seelig J. In vivo proton MR spectroscopy of human gliomas: definition of metabolic coordinates for multi-dimensional classification. Magn Reson Med. 1995;34:242–52. https://doi.org/10.1002/mrm.1910340216.
Heiland DH, Mader I, Schlosser P, Pfeifer D, Carro MS, Lange T, et al. Integrative network-based analysis of magnetic resonance spectroscopy and genome wide expression in glioblastoma multiforme. Sci Rep. 2016;6:29052. https://doi.org/10.1038/srep29052.
Demerath T, Simon-Gabriel CP, Kellner E, Schwarzwald R, Lange T, Heiland DH, et al. Mesoscopic imaging of glioblastomas: are diffusion, perfusion and spectroscopic measures influenced by the radiogenetic phenotype? Neuroradiol J. 2017;30:36–47. https://doi.org/10.1177/1971400916678225.
Diamandis E, Gabriel CPS, Würtemberger U, Guggenberger K, Urbach H, Staszewski O, et al. MR-spectroscopic imaging of glial tumors in the spotlight of the 2016 WHO classification. J Neurooncol. 2018;139:431–40. https://doi.org/10.1007/s11060-018-2881-x.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. https://doi.org/10.1186/gb-2010-11-10-r106.
Hess M, Lenz S, Blaette T, Bullinger L, Binder H. Partitioned learning of deep Boltzmann machines for SNP data. BioRxiv. 2016. https://doi.org/10.1101/095638.
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(835–849):e21. https://doi.org/10.1016/j.cell.2019.06.024.
Acknowledgements
The authors would like to thank Mr. Hansjörg Mast for his assistance in the MR spectroscopy measurements.
Funding
Open Access funding enabled and organized by Projekt DEAL. This is an investigator-initiated study and is being funded by our department’s internal research funding resources. An application for external funding has been submitted to the German Research Foundation (DFG) and is under review.
Author information
Authors and Affiliations
Contributions
PF assisted with patient recruitment, acquisition and analysis of data and wrote most of the manuscript. DHH, IM and HU were in charge of the conception and design of the study, as well as patient recruitment. IM contributed to editing of the manuscript. IM, UW and HU performed analysis of chemical shift imaging. DHH was the main responsible for statistical and genetic analysis and writing parts of the manuscript. DHH and KD developed the algorithm for segmentation. DHH and HU were responsible for submitting the funding proposal. IH was responsible for patient data processing. JB and OS contributed to editing the manuscript. HB was responsible for the conception of the computational analysis. All authors approved the submitted version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study has been approved by the Ethics Committee of the Medical Centre University Freiburg (protocol 360/16_170908) and has been registered at the German Clinical Trials Register (DRKS) under DRKS00019855. Once participants are elected eligible, they will be required to sign two forms of informed consent: the first one for undergoing the imaging and the second one for the extraction of tumour samples for genetic analysis of residual tumour tissue after all clinically relevant investigations requiring said tumour tissue have been completed. The written informed consents will be obtained by treating neurosurgeons as well as by the neuroradiologists. Patients are informed that this study is non-interventional, meaning that they cannot expect a direct benefit from being included in the study, and that they will not undergo additional risks related to the imaging acquisition process or the tumour sample obtainment.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Franco, P., Würtemberger, U., Dacca, K. et al. SPectroscOpic prediction of bRain Tumours (SPORT): study protocol of a prospective imaging trial. BMC Med Imaging 20, 123 (2020). https://doi.org/10.1186/s12880-020-00522-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12880-020-00522-y