
Abstract
Background
In Gram-positive Corynebacterium glutamicum and other members of the suborder Corynebacterianeae, which includes mycobacteria, cell elongation and peptidoglycan biosynthesis is mainly due to polar growth. C. glutamicum lacks an uptake system for the peptidoglycan constituent N-acetylglucosamine (GlcNAc), but is able to catabolize GlcNAc-6-phosphate. Due to its importance in white biotechnology and in order to ensure more sustainable processes based on non-food renewables and to reduce feedstock costs, C. glutamicum strains have previously been engineered to produce amino acids from GlcNAc. GlcNAc also is a constituent of chitin, but it is unknown if C. glutamicum possesses chitinolytic enzymes.
Results
Chitin was shown here not to be growth substrate for C. glutamicum. However, its genome encodes a putative N-acetylglucosaminidase. The nagA 2 gene product was active as β-N-acetylglucosaminidase with 0.27 mM 4-nitrophenyl N,N’-diacetyl-β-D-chitobioside as substrate supporting half-maximal activity. NagA2 was secreted into the culture medium when overproduced with TAT and Sec dependent signal peptides, while it remained cytoplasmic when overproduced without signal peptide. Heterologous expression of exochitinase gene chiB from Serratia marcescens resulted in chitinolytic activity and ChiB secretion was enhanced when a signal peptide from C. glutamicum was used. Colloidal chitin did not support growth of a strain secreting exochitinase ChiB and β-N-acetylglucosaminidase NagA2.
Conclusions
C. glutamicum possesses β-N-acetylglucosaminidase. In the wild type, β-N-acetylglucosaminidase activity was too low to be detected. However, overproduction of the enzyme fused to TAT or Sec signal peptides led to secretion of active β-N-acetylglucosaminidase. The finding that concomitant secretion of endogenous NagA2 and exochitinase ChiB from S. marcescens did not entail growth with colloidal chitin as sole or combined carbon source, may indicate the requirement for higher or additional enzyme activities such as processive chitinase or endochitinase activities.
Similar content being viewed by others


Background
Corynebacteria are Gram-positive microorganisms closely related to mycobacteria. C. glutamicum is considered a model organism for other members of the suborder Corynebacterianeae. Likewise, C. glutamicum is a model bacterium for white biotechnology since it is used for the production of amino acids and derived products [1]. This bacterium can use a variety of carbon sources for growth and production, e.g. sugars (glucose, fructose, sucrose, maltose, ribose), alcohols (ethanol, myo-inositol) and organic acids (acetate, propionate, D- or L-lactate, gluconate, pyruvate) [2–6]. Metabolic engineering has been applied to C. glutamicum aimed at improving amino acid production, to enable production of novel compounds such as diamines [7, 8] or terpenoids [9–11], and to enable a flexible feedstock concept [12]. Feedstock costs are an important part of the overall fermentation costs and access to a wide range of carbon substrates, in particular non-food renewables, is sought [2]. Recombinant strains of C. glutamicum which can utilize glycerol [13, 14], starch [15, 16], arabinose and xylose derived from lignocellulosic biomass [17–21] or the amino sugars glucosamine (GlcN) and N-acetyl-glucosamine (GlcNAc) have been developed [22, 23]. GlcN and GlcNAc represent interesting feedstocks for biotechnological applications since they can be produced by acid hydrolysis of chitin, one of the major waste components generated from shellfish industry [24, 25]. Shrimp farming is a continuously growing sector concentrated in Southeast Asia and China, where every year around 4 million MT of shrimps are harvested, with a chitin-containing waste generation of around 40 % of shrimp dry weight [26, 27]. Thus, the amino sugar fraction of shellfish waste could represent an example of a sustainable, renewable feedstock for industrial fermentations, in particular for nitrogenous compounds such as amino acids and diamines.
C. glutamicum lacks the transporter for GlcNAc so that the heterologous overexpression of a specific transporter is necessary in order to achieve growth on this carbon source [22]. GlcNAc-6P is generated intracellularly during catabolism of N-acetylneuramic acid (Neu5Ac), a carbon source for C. glutamicum. The sialic acid transport and utilization genes are clustered in C. glutamicum ATCC 13032 together with a regulator (cg2936) belonging to the GntR-family transcription factor (in the region cg2929-cg2940) [28, 29]. The genome of C. glutamicum ATCC 13032 contains a gene (cg3158, nagA 2 ) annotated to encode a putative β-N-acetylglucosaminidase precursor, whose activity and function has not yet been studied. β-N-acetylglucosaminidases (E.C. 3.2.1.52) are characterized by the ability to cleave off single units of GlcNAc from the non-reducing ends of short oligosaccharides or peptidoglycan. A common N-acetylglucosaminidase of bacteria is NagZ, a GH3 family hydrolase that participates in bacterial cell wall recycling [30]. In the Gram-negative model organism E. coli, NagZ is located in the cytoplasm and removes GlcNAc from 1,6-anhydroMurNAc-peptides, that derive from the breakdown of the murein envelope by transglycosylases and endopeptidases during growth [31]. In many Gram-negative bacteria, cell wall recycling is also linked to antibiotic response: the organisms can detect β-lactam antibiotics affecting cell wall formation by sensing an increase in intracellular muropeptides concentration, eliciting resistance mechanisms [30].
Whether Gram-positive bacteria recycle their cell wall has been questioned for a long time, but recently putative cell recycling pathways have been elucidated for Bacillus subtilis and Clostridium acetobutylicum [32, 33]. In contrast to E. coli and C. acetobutylicum, muropeptide cleavage occurs extracellularly in B. subtilis, whose NagZ is secreted to the medium, where it hydrolyzes GlcNAc from muropeptide units. The released products are transported into the cytoplasm by specific phosphotransferases and further processed for recycling or energy production [34]. In each generation, up to 50 % of peptidoglycan is turned over in B. subtilis and E. coli [35], due to their cell division mechanism, characterized by intercalation of new cell wall along most of their length. To our knowledge, it is not known whether C. glutamicum recycles peptidoglycan, given that the apical cell elongation occurring during its cell division may not require a massive cell wall breakdown [36, 37]. C. glutamicum lacks orthologs of most of the genes responsible for peptidoglycan recycling as present in E. coli [35]. Moreover, since C. glutamicum does not possess a specific transporter for GlcNAc, it cannot import and utilize GlcNAc as carbon source unless a gene for GlcNAc transport is expressed heterologously [22].
N-acetylglucosaminidase also catalyzes the last step of bacterial chitin degradation. Many chitinolytic organisms produce a cocktail of chitinases that hydrolyze chitin to the disaccharide N,N'-diacetyl chitobiose as major end product, which is eventually cleaved by a chitobiosidase to GlcNAc monomers [38]. GlcNAc production by enzymatic treatment using specific enzymes from chitinolytic organisms and unspecific crude enzymes such as lysozyme, papaine or lipase is seen more favorably than chemical hydrolysis, since enzymatic hydrolysis of chitin operates under mild conditions without generating large amounts of wastes, an environmental concern of the chemical method [24, 39].
Here we report that C. glutamicum possesses N-acetylglucosaminidase activity and that the respective enzyme is encoded by cg3158/nagA 2 . Its activity was compared to that of the already characterized NagZ from B. subtilis. Furthermore, variants of NagA2, fused to different tags for secretion or without secretion signal, were constructed and tested. The variants with higher secreted activity have been combined in a plasmid with a secreted chitinase, ChiB, from the chitinolytic Gram-negative Serratia marcescens, in order to test if the combination of chitinase and N-acetylglucosaminidase activity supports the growth of C. glutamicum with chitin as carbon source.
Results
C. glutamicum ATCC13032 possesses a GH3 N-acetylglucosaminidase
BLAST alignments indicate that nagA 2 encodes a β N-acetylglucosaminidase belonging to the family 3 of glycosidases, which is predicted to be secreted. As other GH3 family β N-acetylglucosaminidases characterized so far, NagA2 possesses the highly conserved sequence motif KHFPGHGX(4) DSH, where the aspartyl and histidyl residues (indicated in bold) form a catalytic dyad as elucidated for the first time for B. subtilis NagZ [40] (Table 1). The gene nagA 2 was cloned in an overexpression vector in order to determine whether it encodes for an active protein. An additional variant of nagA 2 was cloned, replacing the translational start codon GTG with ATG, the preferred start codon of C. glutamicum [41]. Overexpression of the native gene (GTG start codon) led to detectable N-acetylglucosaminidase activity. While most of the activity was found in the cytoplasmic fraction, about one third of the activity was in the supernatant. Replacing the start codon of the nagA 2 gene with ATG led to a two fold increase of the activity both in the cell extract (specific activity of 3.7 ± 0.2 mU mg−1, corresponding to a total activity 0.09 U in a grown 50 mL culture) and in the supernatant fraction (total activity of 0.01 U in the 50 mL broth). By contrast, no extracellular activity was detected upon overexpression of a nagA 2 version lacking the predicted signal peptide, while the intracellular specific activity reached the highest value of 5.4 ± 0.3 mU mg−1, corresponding to about 0.18 U in a grown 50 mL culture (Fig. 1). The overall activity is relatively low when compared to the heterologous overexpression of nagZ from B. subtilis (Fig. 1). NagZ produced in C. glutamicum led to an intracellular specific activity of N-acetylglucosaminidase of 40.7 ± 4.8 mU mg−1 (circa 0.6 U in the 50 mL broth), more than 25 fold higher than for the corresponding C. glutamicum native enzyme. Taken together, the results indicated that nagA 2 encodes a secreted N-acetylglucosaminidase, although secretion was not efficient.

N-Acetylglucosaminidase activities in supernatants (extracellular fraction) and crude extracts (cellular fraction) from cultures of C. glutamicum ΔnanR transformed with the indicated plasmids. Data represent means and SD from activity assays conducted on three independent cultivations. All results were tested for significance using the paired heteroscedastic Student’s t-test. The level of significance of the differences observed between the control and test samples was expressed as one, two or three stars, for * p ≤ 0.05, and ** p ≤ 0.01, respectively. “NS” stands for nonsignificant
Overexpression of nagA 2 fused with homologous N-terminal signal peptides increased extracellular activity of the enzyme
In order to increase the secretion of the enzyme, the putative signal peptide of nagA 2 was replaced by various endogenous signal peptides known to enable secretion in C. glutamicum [42–46]. Different TAT and Sec pathway signal peptides were considered in this work (Table 2): SP0955, a TAT secretion signal homologous to C. glutamicum R cgR_0949 [42, 43]; PS2, a Sec-pathway signal peptide of C. glutamicum ATCC 14067 PS2 protein, which is missing in C. glutamicum ATCC 13032 [44, 45]; PhoD, a TAT-pathway signal peptide from cg2485 encoding secreted alkaline phosphatase [46]; Cmt4 and Cmt1, Sec pathway secretion signals of cg2394 coding for corynomycolyl transferase, and, cg0413 coding for trehalose corynomycolyl transferase, respectively [43].
Both, the overall activity of NagA2 and its secretion efficiency, greatly improved upon replacing the native secretion signal peptide. N-acetylglucosaminidase activity in the supernatant ranged from about 85 % to 93 % of the total activity measured inside the cells and in the broth. The signal peptides SP0955, PhoD and Cmt4 led to the highest enzyme activities in the culture broth (corresponding to 0.29 ± 0.05 U, 0.31 ± 0.02 U and 0.34 ± 0.04 U, respectively).
Thus, about sixty fold more N-acetylglucosaminidase activity as compared to the native enzyme was found in the supernatant (Fig. 1). Signal peptides are cleaved during protein export and are expected not to affect kinetic parameters of the mature enzymes. Indeed, the Km values of NagA2 lacking its own signal peptide (0.266 ± 0.037 mM) or fused to the C. glutamicum PhoD signal peptide (0.270 ± 0.066 mM) were shown to be comparable.
B. subtilis NagZ was also expressed with various signal peptides in C. glutamicum (Fig. 1). However, fusion to endogenous signal peptides did not increase the amount of enzyme activity retrieved in the supernatant. Notably, fusion to the signal peptide from Cg0955 increased intracellular enzyme activity, whereas fusion to signal peptide Cmt4 led to reduced intracellular N-acetylglucosaminidase activity (Fig. 1).
Overexpression of ChiB fused with a secretion signal peptide from C. glutamicum enabled secretion of the enzyme
The heterologous overexpression of chiB from S. marcescens in C. glutamicum resulted in the accumulation of an active product in the cytoplasm. No extracellular activity was detected in a glycol chitin gel assay, indicating that the chiB signal peptide from S. marcescens is not recognized by the host (Fig. 2-I). Secretion was achieved by fusing the sequence encoding the TAT secretion signal peptide from the C. glutamicum gene cg0955 to chiB, and activity was detectable on the glycol chitin gel (Fig. 2-II). Subsequently, chitinase activity was quantified by measuring the hydrolysis of p-nitrophenol from 4-Nitrophenyl N,N’-diacetyl-β-D-chitobioside. C. glutamicum strain ΔnanR (pEKEx3-SP0955-chiB) revealed total chitinase activity of about 0.33 U in the 50 mL culture supernatant. ChiB fusion with the signal peptide from Cg0955 not only allowed secretion, but also increased the specific intracellular activity about eight fold from 0.5 ± 0.1 mU mg−1 for native chiB to 3.8 ± 0.3 mU mg−1 (Table 3).

Dot activity assay on glycol chitin gels to detect chitinase activity. I Five μL of cell extract (A) and supernatant (B) from C. glutamicum WT (pEKEx3) (1) and C. glutamicum WT (pEKEx3-chiB) (2) are spotted on the glycol chitin gel. One μL of purified ChiB from S. marcescens with a specific activity of 28.8 mU mL−1 was spotted as a positive control (C1). ChiB activity is clearly seen in the cell extract of C. glutamicum overexpressing ChiB, while no activity is detected on the supernatant of this strain or in the wildtype. II Triplicates (1–3) of one μL of concentrated (Amicon 30 K) culture supernatant (A) and 5 μL of non-concentrated supernatant (B) of C. glutamicum WT (pEKEx3-SP0955-chiB) and the empty vector control C. glutamicum WT (pEKEx3) (A4) were spotted on the glycol chitin gel
Concomitant overproduction of ChiB and NagA2 is not sufficient to support growth with colloidal chitin in a C. glutamicum strain able to grow with GlcNAc
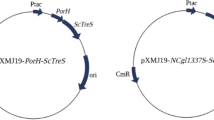
As reported previously, utilization of GlcNAc as substrate in C. glutamicum requires a heterologous GlcNAc uptake system (e.g. NagE from C. glycinophilum) in addition to high NagA and NagB activities [22]. Overexpression of both nagA and nagB is possible either from a plasmid [22] or by derepression via deletion of the GntR-family transcription factor gene nanR, present in the vicinity of the nagAB-scrB operon (Matano & Wendisch, unpublished). In this study, C. glutamicum ΔnanR (pVWEx1-nagE) was transformed with vectors for combined overexpression of endogenous nagA 2 and of chiB from S. marcescens (plasmids maps are depicted in Additional file 1: Figure S1). The chiB gene was cloned with the signal peptide SP0955 and nagA 2 was cloned with the signal peptides SP0955, PhoD or Cmt4. The resulting strains showed a total ChiB activity in the supernatants of around 0.20 U (Table 4). With respect to N-acetylglucosaminidase activities, the strain producing SP0955-nagA 2 showed the highest activity (0.17 ± 0.02 U) as compared to PhoD-nagA 2 (0.08 ± 0.00 U) and Cmt4-nagA 2 (0.11 ± 0.0 U) (Table 4). Therefore, C. glutamicum strain ΔnanR (pVWEx1-nagE) (pEKEx3-SP0955-chiB-SP0955-nagA 2 ) was chosen for growth experiments with colloidal chitin as sole carbon source. While this strain grew with GlcNAc as sole carbon source (Fig. 3a), no growth with colloidal chitin as sole carbon source was observed (data not shown). With minimal medium containing a mixture of 100 mM GlcNAc and 1 % (w/v) colloidal chitin as carbon sources, strain ΔnanR (pVWEx1-nagE) (pEKEx3-SP0955-chiB-SP0955-nagA 2 ) grew to a comparable final OD (25.7 ± 2.6) as with GlcNAc as sole carbon source (final OD of 26.4 ± 0.6) (Fig. 3b). The maximal growth rate was 0.11 ± 0.1 h−1 under both conditions. The control strain ΔnanR (pVWEx1-nagE) reached a comparable final OD (25.7 ± 1.1) in both media, but grew a little slower with a maximal growth rates of 0.085 ± 0.01 h−1 with GlcNAc and 0.88 ± 0.01 h−1 with GlcNAc plus colloidal chitin (Fig. 3).

Growth of C. glutamicum ΔnanR (pVWEx1-nagE) (pEKEx3) [triangles] and ΔnanR (pVWEx1-nagE) (pEKEx3-SP0955-chiB-SP0955-nagA 2 ) [squares] in CgXII minimal medium with 100 mM GlcNAc (a) or 100 mM GlcNAc and 1 % colloidal chitin (b) as carbon and energy sources. Data represent means and SD from three independent cultivations
Thus, although enzyme activities of chitinase ChiB and β-N-acetylglucosaminidase NagA2 could be detected, they might be too low to sustain growth with colloidal chitin. In order to test if indeed colloidal chitin is degraded to GlcNAc, supernatants of strains ∆nanR (pEKEx3-SP0955_chiB-SP0955_nagA2), the control strain WT (pEKEx3), and C. glutamicum WT were analysed for reducing sugars by the 3,5-dinotrosalicidic acid (DNS) colorimetric assay after incubation for 72 h in minimal medium with 1 % colloidal chitin. Indeed, the culture supernatant of strain ∆nanR (pEKEx3-SP0955_chiB-SP0955_nagA2) showed a low (0.88 ± 0.08 mM), but significantly (p < 0.01) higher concentration of reducing sugars than the supernatants of the control strain WT (pEKEx3) and WT (0.72 ± 0.01 mM and 0.70 ± 0.05 mM, respectively, Fig. 4). Despite of some degree of colloidal degradation, the amount of released GlcNAc appeared insufficient to promote cell growth.

Quantification of GlcNAc in culture supernatant by DNS colorimetric assay in WT, WT (pEKEx3) and ∆nanR (pEKEx3-SP0955-chiB-SP0955-nagA2). Values and error bars represent the mean and the standard errors of triplicate cultures. All results were tested for significance using the paired heteroscedastic Student’s t-test. The level of significance of the differences observed between the control and test samples was expressed as one, two or three stars, for * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001, respectively. “NS” stands for nonsignificant
Discussion
Here, C. glutamicum was shown to possess N-acetylglucosaminidase activity that is encoded by cg3158/nagA 2 . Structurally, the NagA2 protein belongs to the family 3 glycoside hydrolases, and among these, the N-acetyl-β-D-glucosaminidases show a selective specificity for GlcNAc as substrate [47] with only few exceptions [48]. N-acetyl-β-D-glucosaminidase activity was assayed with 4-nitrophenyl N,N’-diacetyl-β-D-chitobioside as substrate, and about 0.27 mM supported half-maximal activity. In comparison, NagZ from E. coli had a higher Km on the same substrate (0.43 mM) [49], whereas NagZ of B. subtilis showed an about two fold lower Km of 0.11 ± 0.0 mM with 4'-methylumbelliferyl-β-GlcNAc as substrate [40].
The role of the NagA2 activity in C. glutamicum is still unclear. Analysis of the C. glutamicum transcriptome revealed that the nagA2 gene is transcribed as leaderless transcript with a relatively low RNA abundance [41]. It is not known whether nagA2 expression is regulated in C. glutamicum. The adjacent gene cg3157 encodes a hitherto uncharacterized protein with VanW (pfam04294) and peptidoglycan binding domains (pfam12229) that may play a role in cell wall biosynthesis. In contrast, the orthologous nagZ is part of operon in E. coli (hinT-ycfL-lpoB-thiK-nagZ-ycfP) and B. subtilis (ybbIHFEDC). NagZ from B. subtilis and E. coli have been shown to play important roles in cell wall recycling [30, 32, 50]. C. glutamicum lacks orthologs for most of the genes responsible for muropeptide recycling in B. subtilis and orthologs for E. coli genes necessary for import and catabolism of anhydromuropeptides. Therefore, further studies need to be performed in order to elucidate whether C. glutamicum has a peptidoglycan recycling mechanism and if NagA2 is involved.
The endogenous protein NagA2 was shown to be secreted, although inefficiently. Increased protein levels due to changing the nagA 2 translational start codon from GTG to the more common ATG increased total activity. But even though total activity doubled, only around 10 % was found in the supernatant, showing that the endogenous NagA2 signal peptide supports secretion rather inefficiently.
Protein secretion has been studied for decades in C. glutamicum and recently a strain exhibiting potential as host for industrial-scale production of recombinant proteins has been commercialized as Corynex™. C. glutamicum is a favorable host for protein production and secretion, i.e. because it lacks extracellular proteolytic activities [51]. However, shortcomings such as its mycolic acid layer that in combination with the underlying peptidoglycan-arabinogalactan layer constitutes a second permeability barrier [52, 53] or an additional S-layer (present only in some strains) can hamper its utilization for protein production [54]. The strains used in this study are based on ATCC 13032, which lacks an S-layer [44]. C. glutamicum is not widely used for protein production, which may in part be due to variable yields depending on the target protein and on the target signal [42, 54–56].
The promoter and signal sequence of the cspB gene encoding PS2, the major protein secreted by the organism, have been used in many studies of heterologous protein secretion in C. glutamicum [43, 44, 57, 58]. A systematic screen of signal peptides for secretion of α-amylase from Geobacillus stearothermophilus in C. glutamicum showed up to 150-fold better secretion using signal peptides from e.g. genes cg0955, cmt1 and cmt4, and phoD when compared to fusions with the PS2 secretion signal [43]. Conversely, secretion of endogenous NagA2 as well as heterologous ChiB fused to these signal peptides did not differ much in terms of overall secreted activity, utilizing different secretion signals (Fig. 1). Notably, signal peptides involved in the Sec pathway, (PS2, Cmt1 and Cmt4) or the Tat pathway (Cg0955 and PhoD) functioned similarly with NagA2 and ChiB (Fig. 1), whereas for other proteins, e.g. GFP, the translocation efficiency and/or final protein activity was strongly affected by the route of transport [59–61].
Despite of detection of GlcNAc units in culture supernatant, secretion of ChiB and NagA2 by C. glutamicum strain ΔnanR (pVWEx1-nagE) (pEKEx3-SP0955-chiB-SP0955-nagA 2 ) was too low to support growth on colloidal chitin. Extracellular activities were never higher than 5 mU mL−1 (Fig. 1). This contrasts to recombinant C. glutamicum strains which are able to grow with starch as sole carbon source due to secretion of heterologous α-amylases [15, 57] due to about 100 fold higher α-amylase activities (400 to 650 mU mL−1).
A strategy for the hydrolysis of complex bio-polymers, alternative to the secretion of degrading enzymes, is their display on the cell surface, anchored to membrane integral proteins. One notable example for this approach is the heterologous expression of the α-amylase from Streptococcus bovis 148 (amyA), that allowed the growth with starch as carbon source when fused to C. glutamicum anchor proteins such as porins [62], the glutamate exporter NCgl1221 [63], or heterologous proteins such as PgsA from B. subtilis [16]. More recently, it was shown that the combined surface display in C glutamicum of endoglucanase [64] and β-glucosidase (BglA) from Clostridium thermocellum, anchored to the mechanosensitive channel Msc, enabled the saccharification of lignocellulosic material, leading to up to 6-fold increased reducing sugar generation when compared to secreted cellulases [65]. It remains to be established if surface display of N-acetylglucosaminidases and chitinases is superior to secretion of these enzymes. It may also be necessary to simultaneously overproduce chitinases with different mechanisms of action, e.g. combining an endo-chitinase, e.g. ChiC from S. marcescens, with a processive chitinase, e.g. ChiB from S. marcescens [24, 66, 67].
Conclusion
C. glutamicum possesses β-N-acetylglucosaminidase. In the wild type, β-N-acetylglucosaminidase activity was too low to be detected. However, overproduction of the enzyme fused to TAT or Sec signal peptides led to secretion of active β-N-acetylglucosaminidase. The engineering of the signal peptide improved the secretion more than 60 folds as compared to the native sequence. NagZ of B. subtilis could be overproduced, but was not excreted efficiently, even when fused to C. glutamicum TAT or Sec signal peptides. Concomitant secretion of exochitinase ChiB from S. marcescens and endogenous NagA2 did not result in growth with colloidal chitin as sole or combined carbon source. This may indicate that either higher enzyme activities are needed or that additional enzymes such as processive chitinase or endochitinase activities may have to be overproduced.
Methods
Microorganisms, media, and cultivation conditions
The strains and plasmids used are listed in Table 5. The standard medium Luria–Bertani (LB) was used for E. coli [68], and brain heart infusion medium (BHI; Difco) was applied as complex medium for C. glutamicum. CgXII was used as minimal medium [69], supplemented with 100 mM N-acetyl-glucosamine and/or 100 mM N-acetyl-glucosamine, plus 10 g L−1 of colloidal chitin as carbon source. When appropriate, spectinomycin (100 mg L−1), kanamycin (25 mg L−1) and isopropyl-β-D-thiogalactopyranoside (IPTG, 0.1 mM) were added. E. coli was grown at 37 °C and C. glutamicum at 30 °C in 500 or 100 ml Erlenmeyer flask with 50 ml or 20 ml medium, respectively, on a rotary shaker at 120 rpm. Bacterial growth in liquid cultures was followed by measuring the optical density at 600 nm (OD600).
Preparation of colloidal chitin
A modified [70] method was used for colloidal chitin preparation. 5 g of chitin (Sigma) were added to 50 mL of concentrated hydrochloric acid. The mixture was incubated overnight at 4 °C while continuously stirring. Chitin was precipitated as colloidal suspension by the slow addition of deionized water and the chitin suspension was washed with deionized water until a neutral pH value was reached. The colloidal chitin obtained was resuspended in medium for growth assay in a final concentration of 10 g L−1.
DNA preparation, manipulation and transformation
Plasmids were constructed in E. coli DH5α from PCR-generated fragments (KOD Hot Start DNA Polymerase; Novagen), using C. glutamicum ATCC 13032, S. marcescens ATCC 13880 genomic DNA as template. C. glutamicum genomic DNA was prepared as described [71]; S. marcescens ATCC 13880 (DSM 30121) genomic DNA was obtained by DSMZ German Collection of Microorganism and Cell Cultures (Braunschweig, Germany). A nagZ gene (GenBank Acc. No. CAB11942.1) from B. subtilis 168 (ATCC 23857) was synthesized in a codon optimized version for overexpression in E. coli (Genscript, USA). The oligonucleotides used in this study are listed in (Additional file 1: Table S1). E. coli was transformed by standard methods [68] and the plasmids were introduced by electroporation into C. glutamicum strains as described [69]. Transformants containing two compatible plasmids, here pVWEx1 and pEKEx3 and their derivatives, were selected by plating on agar plates containing kanamycin (25 μg/ml) and spectinomycin (100 μg/ml). Vector maps of pVWEx1-nagE and pEKEx3-SP0955-chiB-SP0955-nagA2 are shown in Additional file 1: Figure S1. Transformants were analysed by PCR with appropriate primers; the absence of mutations in the cloned genes was verified by sequencing.
Construction of plasmids and strains
For the construction of plasmids expressing chiB, nagZ and nagA 2 , with their own signal peptide or in fusion with C. glutamicum signal peptides, KOD PCR products were ligated into BamHI, EcoRI digested pEKEx3 via Gibson assembly [72]. The chiB gene was cloned into pEKEx3 in its native form and in fusion with a signal peptide from gene cg0955 of C. glutamicum ATCC 13032 (hence SP0955). The gene nagA 2 has been cloned in its native form, including its own signal peptide, with a mutation on the first triplet replacing the original GTG start codon with the ATG triplet, without its own putative signal sequence (hence SP-less- nagA 2 ) and with its signal peptide replaced by secretion sequences from C. glutamicum, here named SP0955, PS2, PhoD, Cmt4 and Cmt1.
B. subtilis nagZ has been cloned with its own signal peptide or in a signal peptide-less form, in fusion with the above mentioned signal peptides SP0955, PS2, PhoD, Cmt4 and Cmt1.
Glycol chitin preparation and dot blot chitinase assay
Glycol chitin was prepared by re-acetylation of glycol chitosan. 1 g of glycol chitosan (Sigma-Aldrich) was added to 20 mL of 10 % acetic acid and the mixture was incubated overnight while continuously stirring. 90 mL of methanol were slowly added and the solution was vacuum filtered on Buchner funnel. 1.5 mL of acetic anhydride were added to the filtrate and the solution was incubated 30 min at room temperature allowing the formation of a solid gel. The gel was cut into small pieces and homogenized in a waring blender. The homogenized gel was centrifuged and washed with methanol and resuspended a last time in 100 ml H2O to give a ca 1 % (w/v) solution of glycol chitin.
A dot blot assay on 12 % (v/v) acrylamide gel, with 50 mM TEA buffer (pH 7.0), containing 0.01 % (v/v) of glycol chitin was used for chitinolytic activity determination. Fractions of cell extract or supernatant from C. glutamicum WT (pEKEx3-chiB), C. glutamicum WT (pEKEx3) and ΔnanR (pEKEx3 SP0955-chiB) were spotted on the gel. After overnight incubation the gel was stained for 30 minutes with 0.01 % (w/v) Calcofluor White M2R (Sigma Aldrich, Germany) solution, destained and visualized by UV illumination [73]. Chitinolytic activity is revealed as dark halo on fluorescent background.
Chitinase and N-acetylglucosaminidase activity assays
Aliquots from C. glutamicum BHI cultures were withdrawn during the exponential growth phase and cells where harvested by centrifugation (10 min, 3,200 x g and 4 °C). Supernatant and cell extract fractions from the aliquots were used for chitinase and N-acetylglucosaminidase activity quantification. Cells were disrupted by ultrasonic treatment (UP 200S; Dr. Hielscher GmbH, Teltow, Germany) with an amplitude of 50 % and a duty circle of 0.5 for 7 min, upon resuspension in 100 mM phosphate buffer (pH 7.0). The cell suspension was centrifuged for 1 h at 4 °C and 16,000 rpm and the soluble cell extract was recovered. Exo-chitinase and N-acetylglucosaminidase activity were determined by measuring the hydrolysis of p-nitrophenol from 4-Nitrophenyl N,N’-diacetyl-β-D-chitobioside and 4-Nitrophenyl N-acetyl-β-D-glucosaminide (Sigma Aldrich, Germany), respectively. The substrates were dissolved in 100 mM phosphate buffer (pH 7.0) solution at a concentration of 1 mg mL−1 and the activity was determined, following the chitinases assay protocol from Sigma Aldrich in 96 well plates, measuring the absorption at 405 nm of hydrolyzed p-nitrophenol upon addition of sodium carbonate stop solution. Km values have been estimated with the Eadie Hofstee plot for NagA2 without its own signal peptide and as fusion protein with PhoD signal peptide, measuring the activity with different concentration of 4-Nitrophenyl N,N’-diacetyl-β-D-chitobioside in the cell extract fraction and in the supernatant, respectively.
Quantification of GlcNAc via DNS colorimetric assay
GlcNAc released in the supernatant was determined spectrophotometrically by the Dinitrosalicylic acid (DNS) method following the protocol described by Miller [74] at 540 nm using 3 ml of culture supernatant. Concentrations of the released sugars were estimated using a standard calibration curve of GlcNAc (Sigma Aldrich, Germany) in a range of 0–2 mM.
References
Wendisch VF. Microbial production of amino acids and derived chemicals: synthetic biology approaches to strain development. Curr Opin Biotechnol. 2014;30C:51–8.
Blombach B, Seibold GM. Carbohydrate metabolism in Corynebacterium glutamicum and applications for the metabolic engineering of L-lysine production strains. Appl Microbiol Biotechnol. 2010;86:1313–22.
Stansen C, Uy D, Delaunay S, Eggeling L, Goergen JL, Wendisch VF. Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Environ Microbiol. 2005;71:5920–8.
Krings E, Krumbach K, Bathe B, Kelle R, Wendisch VF, Sahm H, Eggeling L. Characterization of myo-inositol utilization by Corynebacterium glutamicum: the stimulon, identification of transporters, and influence on L-lysine formation. J Bacteriol. 2006;188:8054–61.
Claes WA, Puhler A, Kalinowski J. Identification of two prpDBC gene clusters in Corynebacterium glutamicum and their involvement in propionate degradation via the 2-methylcitrate cycle. J Bacteriol. 2002;184:2728–39.
Arndt A, Eikmanns BJ. Regulation of carbon metabolism in Corynebacterium glutamicum. In: Burkovski A, editor. Corynebacteria: genomics and molecuar biology. Norfolk: Caister Acadeic Press; 2008. p. 155–82.
Schneider J, Eberhardt D, Wendisch VF. Improving putrescine production by Corynebacterium glutamicum by fine-tuning ornithine transcarbamoylase activity using a plasmid addiction system. Appl Microbiol Biotechnol. 2012;95:169–78.
Schneider J, Wendisch VF. Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2010;88:859–68.
Heider SA, Peters-Wendisch P, Netzer R, Stafnes M, Brautaset T, Wendisch VF. Production and glucosylation of C50 and C 40 carotenoids by metabolically engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2014;98:1223–35.
Frohwitter J, Heider SA, Peters-Wendisch P, Beekwilder J, Wendisch VF. Production of the sesquiterpene (+)-valencene by metabolically engineered Corynebacterium glutamicum. J Biotechnol. 2014;191:205–13.
Heider SA, Peters-Wendisch P, Wendisch VF. Carotenoid biosynthesis and overproduction in Corynebacterium glutamicum. BMC Microbiol. 2012;12:198.
Zahoor A, Lindner SN, Wendisch VF. Metabolic engineering of Corynebacterium glutamicum aimed at alternative carbon sources and new products. Comput Struct Biotechnol J. 2012;3:e201210004.
Meiswinkel TM, Rittmann D, Lindner SN, Wendisch VF. Crude glycerol-based production of amino acids and putrescine by Corynebacterium glutamicum. Bioresour Technol. 2013;145:254–8.
Rittmann D, Lindner SN, Wendisch VF. Engineering of a glycerol utilization pathway for amino acid production by Corynebacterium glutamicum. Appl Environ Microbiol. 2008;74:6216–22.
Seibold G, Auchter M, Berens S, Kalinowski J, Eikmanns BJ. Utilization of soluble starch by a recombinant Corynebacterium glutamicum strain: growth and lysine production. J Biotechnol. 2006;124:381–91.
Tateno T, Fukuda H, Kondo A. Production of L-Lysine from starch by Corynebacterium glutamicum displaying alpha-amylase on its cell surface. Appl Microbiol Biotechnol. 2007;74:1213–20.
Kawaguchi H, Sasaki M, Vertes AA, Inui M, Yukawa H. Engineering of an L-arabinose metabolic pathway in Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2008;77:1053–62.
Kawaguchi H, Vertes AA, Okino S, Inui M, Yukawa H. Engineering of a xylose metabolic pathway in Corynebacterium glutamicum. Appl Environ Microbiol. 2006;72:3418–28.
Gopinath V, Meiswinkel TM, Wendisch VF, Nampoothiri KM. Amino acid production from rice straw and wheat bran hydrolysates by recombinant pentose-utilizing Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2011;92:985–96.
Meiswinkel TM, Gopinath V, Lindner SN, Nampoothiri KM, Wendisch VF. Accelerated pentose utilization by Corynebacterium glutamicum for accelerated production of lysine, glutamate, ornithine and putrescine. Microb Biotechnol. 2013;6:131–40.
Schneider J, Niermann K, Wendisch VF. Production of the amino acids L-glutamate, L-lysine, L-ornithine and L-arginine from arabinose by recombinant Corynebacterium glutamicum. J Biotechnol. 2011;154:191–8.
Matano C, Uhde A, Youn JW, Maeda T, Clermont L, Marin K, Kramer R, Wendisch VF, Seibold GM. Engineering of Corynebacterium glutamicum for growth and L-lysine and lycopene production from N-acetyl-glucosamine. Appl Microbiol Biotechnol. 2014;98:5633–43.
Uhde A, Youn JW, Maeda T, Clermont L, Matano C, Kramer R, Wendisch VF, Seibold GM, Marin K. Glucosamine as carbon source for amino acid-producing Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2013;97:1679–87.
Chen JK, Shen CR, Liu CL. N-Acetylglucosamine: production and applications. Mar Drugs. 2010;8:2493–516.
Wang SL, Chang TJ, Liang TW. Conversion and degradation of shellfish wastes by Serratia sp. TKU016 fermentation for the production of enzymes and bioactive materials. Biodegradation. 2010;21:321–33.
Kandra P, Challa MM, Jyothi HKP. Efficient use of shrimp waste: present and future trends. Appl Microbiol Biotechnol. 2012;93:17–29.
Global Aquaculture Production [http://www.fao.org/fishery/statistics/global-aquaculture-production/en]. Accessed 30 Oct 2014.
Gruteser N, Marin K, Kramer R, Thomas GH. Sialic acid utilization by the soil bacterium Corynebacterium glutamicum. FEMS Microbiol Lett. 2012;336:131–8.
Holder JW, Ulrich JC, DeBono AC, Godfrey PA, Desjardins CA, Zucker J, Zeng Q, Leach AL, Ghiviriga I, Dancel C, et al. Comparative and functional genomics of Rhodococcus opacus PD630 for biofuels development. PLoS Genet. 2011;7:e1002219.
Johnson JW, Fisher JF, Mobashery S. Bacterial cell-wall recycling. Ann N Y Acad Sci. 2013;1277:54–75.
Park JT, Uehara T. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol Mol Biol Rev. 2008;72:211–27.
Litzinger S, Duckworth A, Nitzsche K, Risinger C, Wittmann V, Mayer C. Muropeptide rescue in Bacillus subtilis involves sequential hydrolysis by beta-N-acetylglucosaminidase and N-acetylmuramyl-L-alanine amidase. J Bacteriol. 2010;192:3132–43.
Reith J, Mayer C. Characterization of a glucosamine/glucosaminide N-acetyltransferase of Clostridium acetobutylicum. J Bacteriol. 2011;193:5393–9.
Reizer J, Saier Jr MH, Deutscher J, Grenier F, Thompson J, Hengstenberg W. The phosphoenolpyruvate:sugar phosphotransferase system in gram-positive bacteria: properties, mechanism, and regulation. Critic Rev Microbiol. 1988;15:297–338.
Reith J, Mayer C. Peptidoglycan turnover and recycling in Gram-positive bacteria. Appl Microbiol Biotechnol. 2011;92:1–11.
Letek M, Fiuza M, Ordonez E, Villadangos AF, Ramos A, Mateos LM, Gil JA. Cell growth and cell division in the rod-shaped actinomycete Corynebacterium glutamicum. Anton Leeuw Int J G. 2008;94:99–109.
Donovan C, Bramkamp M. Cell division in Corynebacterineae. Front Microbiol. 2014;5:132.
Vaaje-Kolstad G, Horn SJ, Sorlie M, Eijsink VG. The chitinolytic machinery of Serratia marcescens-a model system for enzymatic degradation of recalcitrant polysaccharides. FEBS J. 2013;280:3028–49.
Sashiwa H, Fujishima S, Yamano N, Kawasaki N, Nakayama A, Muraki E, Hiraga K, Oda K, Aiba S. Production of N-acetyl-D-glucosamine from alpha-chitin by crude enzymes from Aeromonas hydrophila H-2330. Carbohydr Res. 2002;337:761–3.
Litzinger S, Fischer S, Polzer P, Diederichs K, Welte W, Mayer C. Structural and kinetic analysis of Bacillus subtilis N-acetylglucosaminidase reveals a unique Asp-His dyad mechanism. J Biol Chemi. 2010;285:35675–84.
Pfeifer-Sancar K, Mentz A, Ruckert C, Kalinowski J. Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genomics. 2013;14:888.
Teramoto H, Watanabe K, Suzuki N, Inui M, Yukawa H. High yield secretion of heterologous proteins in Corynebacterium glutamicum using its own Tat-type signal sequence. Appl Microbiol Biotechnol. 2011;91:677–87.
Watanabe K, Tsuchida Y, Okibe N, Teramoto H, Suzuki N, Inui M, Yukawa H. Scanning the Corynebacterium glutamicum R genome for high-efficiency secretion signal sequences. Microbiology. 2009;155:741–50.
Hansmeier N, Albersmeier A, Tauch A, Damberg T, Ros R, Anselmetti D, Puhler A, Kalinowski J. The surface (S)-layer gene cspB of Corynebacterium glutamicum is transcriptionally activated by a LuxR-type regulator and located on a 6 kb genomic island absent from the type strain ATCC 13032. Microbiol. 2006;152:923–35.
Hansmeier N, Bartels FW, Ros R, Anselmetti D, Tauch A, Puhler A, Kalinowski J. Classification of hyper-variable Corynebacterium glutamicum surface-layer proteins by sequence analyses and atomic force microscopy. J Biotechnol. 2004;112:177–93.
Meissner D, Vollstedt A, van Dijl JM, Freudl R. Comparative analysis of twin-arginine (Tat)-dependent protein secretion of a heterologous model protein (GFP) in three different Gram-positive bacteria. Appl Microbiol Biotechnol. 2007;76:633–42.
Chitlaru E, Roseman S. Molecular cloning and characterization of a novel beta-N-acetyl-D-glucosaminidase from Vibrio furnissii. J Biol Chem. 1996;271:33433–9.
Mayer C, Vocadlo DJ, Mah M, Rupitz K, Stoll D, Warren RA, Withers SG. Characterization of a beta-N-acetylhexosaminidase and a beta-N-acetylglucosaminidase/beta-glucosidase from Cellulomonas fimi. The FEBS J. 2006;273:2929–41.
Yem DW, Wu HC. Purification and properties of beta-N-acetylglucosaminidase from Escherichia coli. J Bacteriol. 1976;125:324–31.
Jacobs C, Frere JM, Normark S. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in gram-negative bacteria. Cell. 1997;88:823–32.
Billman-Jacobe H, Wang L, Kortt A, Stewart D, Radford A. Expression and secretion of heterologous proteases by Corynebacterium glutamicum. Appl Environ Microbiol. 1995;61:1610–3.
Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci U S A. 2008;105:3963–7.
Marchand CH, Salmeron C, Raad RB, Méniche X, Chami M, Masi M, Bayan N. Biochemical disclosure of the mycolate outer membrane of Corynebacterium glutamicum. J Bacteriol. 2012;194:587–97.
Matsuda Y, Itaya H, Kitahara Y, Theresia NM, Kutukova EA, Yomantas YA, Date M, Kikuchi Y, Wachi M. Double mutation of cell wall proteins CspB and PBP1a increases secretion of the antibody Fab fragment from Corynebacterium glutamicum. Microb Cell Fact. 2014;13:56.
Date M, Itaya H, Matsui H, Kikuchi Y. Secretion of human epidermal growth factor by Corynebacterium glutamicum. Lett Appl Microbiol. 2006;42:66–70.
Date M, Yokoyama K, Umezawa Y, Matsui H, Kikuchi Y. Production of Native-Type Streptoverticillium mobaraense Transglutaminase in Corynebacterium glutamicum. Appl Environ Microbiol. 2003;69:3011–4.
Tateno T, Fukuda H, Kondo A. Direct production of L-lysine from raw corn starch by Corynebacterium glutamicum secreting Streptococcus bovis alpha-amylase using cspB promoter and signal sequence. Appl Microbiol Biotechnol. 2007;77:533–41.
Kikuchi Y, Date M, Yokoyama K, Umezawa Y, Matsui H. Secretion of active-form Streptoverticillium mobaraense transglutaminase by Corynebacterium glutamicum: Processing of the pro-transglutaminase by a cosecreted subtilisin-like protease from Streptomyces albogriseolus. Appl Environ Microb. 2003;69:358–66.
Feilmeier BJ, Iseminger G, Schroeder D, Webber H, Phillips GJ. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol. 2000;182:4068–76.
Santini CL, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu LF. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. The J Biol Chem. 2001;276:8159–64.
Thomas JD, Daniel RA, Errington J, Robinson C. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol. 2001;39:47–53.
Tateno T, Hatada K, Tanaka T, Fukuda H, Kondo A. Development of novel cell surface display in Corynebacterium glutamicum using porin. Appl Microbiol Biotechnol. 2009;84:733–9.
Yao W, Chu C, Deng X, Zhang Y, Liu M, Zheng P, Sun Z. Display of alpha-amylase on the surface of Corynebacterium glutamicum cells by using NCgl1221 as the anchoring protein, and production of glutamate from starch. Arch Microbiol. 2009;191:751–9.
Polena V, Mergui JL, Perrot N, Poncelet C, Barranger E, Uzan S. Long-term results of hysteroscopic myomectomy in 235 patients. Eur J Obstet Gynecol Reprod Biol. 2007;130:232–7.
Kim SJ, Hyeon JE, Jeon SD, Choi GW, Han SO. Bi-functional cellulases complexes displayed on the cell surface of Corynebacterium glutamicum increase hydrolysis of lignocelluloses at elevated temperature. Enzyme Microb Technol. 2014;66:67–73.
Horn SJ, Sørlie M, Vaaje-Kolstad G, Norberg AL, Synstad B, Vårum KM, Eijsink VGH. Comparative studies of chitinases A, B and C from Serratia marcescens. Biocatal Biotransformation. 2006;24:39–53.
Horn SJ, Sikorski P, Cederkvist JB, Vaaje-Kolstad G, Sorlie M, Synstad B, Vriend G, Varum KM, Eijsink VG. Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc Natl Acad Sci U S A. 2006;103:18089–94.
Sambrook J, Russell D. Molecular cloning. A laboratory manual. 3rd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001.
Eggeling L, Reyes O. Experiments. In: Eggeling L, Bott M, editors. Handbook of Corynebacterium glutamicum. Boca Raton: CRC Press; 2005.
Hsu SC, Lockwood JL. Powdered Chitin Agar as a selective medium for enumeration of actinomycetes in water and soil. Appl Microbiol. 1975;29:422–6.
Lambert C, Erdmann A, Eikmanns M, Kramer R. Triggering glutamate excretion in Corynebacterium glutamicum by modulating the membrane state with local anesthetics and osmotic gradients. Appl Environ Microbiol. 1995;61:4334–42.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison 3rd CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–5.
Trudel J, Asselin A. Detection of chitinase activity after polyacrylamide gel electrophoresis. Anal Biochem. 1989;178:362–6.
Miller GL. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem. 1959;31:426–8.
Grant SG, Jessee J, Bloom FR, Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci U S A. 1990;87:4645–9.
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H, Eikmanns BJ. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol. 2001;3:295–300.
Acknowledgements
None.
Funding
Work by CM and VFW was funded in part by grants 0315589G from BMBF in the CRP “Corynebacterium: improving flexibility and fitness for industrial production”. We acknowledge support of the publication fee by Deutsche Forschungsgemeinschaft and the Open Access Publication Funds of Bielefeld University.
Availability of data and material
All data are provided in the main manuscript and the supplementary files.
Authors’ contributions
VFW, BM and CM designed the experiments, reviewed and revised the manuscript. SK and SH supplied the nagZ gene from B. subtilis 168 used in the study and provided support with the analytical methods for determining the activity of chitin degrading enzymes. ES planned and conduct some experiments. CM conducted the experiments, analyzed the results and drafted the manuscript. VFW coordinated the study and helped finalize the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Table S1. Oligonucleotides used in this study. Figure S1 Plasmid maps of pVWEx1-nagE [A] and pEKEx3-SP0955-chiB-SP0955-nagA2 [B]. Functional elements of the plasmid pVWEx1-nagE include antibiotic markers (Km, kanamycin), origin of replication (pHM1519), nagE (GlcNAc-specific PTS from Corynebacterium glycinophilum DSM45794). Functional elements of the plasmid pEKEx3-SP0955-chiB-SP0955-nagA2 include relevant restriction sites, antibiotic markers (Spec, spectomycin), origin of replication (pBL1), tat (SP0955), chiB (chitinase B, Serratia marcescens), nagA2 (β-N-acetylglucosaminidase, C. glutamicum). (DOCX 246 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Matano, C., Kolkenbrock, S., Hamer, S.N. et al. Corynebacterium glutamicum possesses β-N-acetylglucosaminidase. BMC Microbiol 16, 177 (2016). https://doi.org/10.1186/s12866-016-0795-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-016-0795-3