
Abstract
Background
Campylobacter jejuni is a major cause of human gastroenteritis yet there is limited knowledge of how disease is caused. Molecular genetic approaches are vital for research into the virulence mechanisms of this important pathogen. Vectors that allow expression of genes in C. jejuni via recombination onto the chromosome are particularly useful for genetic complementation of insertional knockout mutants and more generally for expression of genes in particular C. jejuni host backgrounds.
Methods
A series of three vectors that allow integration of genes onto the C. jejuni chromosome were constructed by standard cloning techniques with expression driven from three different strong promoters. Following integration onto the C. jejuni chromosome expression levels were quantified by fluorescence measurements and cells visualized by fluorescence microscopy.
Results
We have created plasmid, pCJC1, designed for recombination-mediated delivery of genes onto the C. jejuni chromosome. This plasmid contains a chloramphenicol resistance cassette (cat) with upstream and downstream restriction sites, flanked by regions of the C. jejuni pseudogene Cj0223. Cloning of genes immediately upstream or downstream of the cat gene allows their subsequent introduction onto the C. jejuni chromosome within the pseudogene. Gene expression can be driven from the native gene promoter if included, or alternatively from the cat promoter if the gene is cloned downstream of, and in the same transcriptional orientation as cat. To provide increased and variable expression of genes from the C. jejuni chromosome we modified pCJC1 through incorporation of three relatively strong promoters from the porA, ureI and flaA genes of C. jejuni, Helicobacter pylori and Helicobacter pullorum respectively. These promoters along with their associated ribosome binding sites were cloned upstream of the cat gene on pCJC1 to create plasmids pCJC2, pCJC3 and pCJC4. To test their effectiveness, a green fluorescent protein (gfp) reporter gene was inserted downstream of each of the three promoters and following integration of promoter-gene fusions onto the C. jejuni host chromosome, expression levels were quantified. Expression from the porA promoter produced the highest fluorescence, from flaA intermediate levels and from ureI the lowest. Expression of gfp from the porA promoter enabled visualization by fluorescent microscopy of intracellular C. jejuni cells following invasion of HeLa cells.
Conclusions
The plasmids constructed allow stable chromosomal expression of genes in C. jejuni and, depending on the promoter used, different expression levels were obtained making these plasmids useful tools for genetic complementation and high level expression.
Similar content being viewed by others

Background
Campylobacters, predominantly Campylobacter jejuni and Campylobacter coli, are the most commonly reported causal agents of zoonotic infection with an estimated 400–500 million cases annually worldwide [1]. Campylobacteriosis is an acute, generally self-limiting, diarrhoeal disease [2, 3] with a number of infrequent but serious sequelae of infection including the peripheral neuropathy Guillain-Barre syndrome. Due to its importance as a human pathogen there are many research groups employing molecular genetic approaches to investigate C. jejuni virulence and transmission. However, the availability of tractable genetic tools is relatively limited and our knowledge of how C. jejuni causes disease is thus also limited compared to other enteropathogens such as Escherichia coli and Salmonella species.
One of the key methods for investigating C. jejuni gene function is inactivation of specific genes through insertion of antibiotic (generally kanamycin or chloramphenicol) resistance cassettes via recombination-mediated allelic replacement [4, 5]. To confirm associated phenotypes such mutants are often genetically complemented with at least partial restoration of phenotype confirming the specific role of the gene product under investigation. The two approaches for complementation in C. jejuni are reintroduction of a functional gene either on a replicating plasmid [6, 7] or through chromosomal integration [8]. The latter method involves construction of suicide plasmids that allow recombination-driven introduction of functional genes at specific loci on the C. jejuni chromosome. These loci are chosen so as to minimise potential for unwanted effects of insertion and include intergenic region of rRNA genes [8], or pseudogenes such as Cj0046 [9, 10] and Cj0223 [11].
An important consideration when designing genetic complementation strategies is the nature of the promoter driving gene expression. Ideally one would use the promoter region from which the gene is expressed [10], but these are often difficult to identify accurately and if the gene is part of an operon, may be located some distance away potentially requiring further cloning steps. A simpler and more commonly used strategy is to use the promoter associated with the antibiotic resistance cassette and insert the gene immediately downstream without an intervening transcriptional terminator. However other more or less well characterized promoters from C. jejuni such as that of the iron induced gene fdxA have also been employed [12].
The aim of this study was to generate plasmids for integrating selected genes onto the C. jejuni chromosome that allow expression at different levels. To this end we employed one of three distinct Campylobacter or Helicobacter promoter regions to drive gene expression. Using these plasmids to introduce reporter gene gfp onto the C. jejuni NCTC 11168 chromosome we measured three significantly different levels of gene expression. The highest of these enabled us for the first time to readily visualize intracellular fluorescent C. jejuni cells expressing chromosomal gfp during in vitro cell invasion experiments using a standard fluorescent microscope. The use of these plasmids to generate stable highly fluorescent C. jejuni strains should have widespread applications and improve our knowledge of the virulence and transmission of this major pathogen. These plasmids will also be useful for both genetic complementation and more generally for expressing genes of other origin in C. jejuni backgrounds.
Methods
Bacterial strains
Escherichia coli XL10 gold strains (Stratagene) were grown in Luria-Bertani (LB) broth or on LB agar plates. C. jejuni NCTC 11168 and H. pullorum NCTC 12824 strains from the UK National Collection of Type Cultures were grown on Columbia agar containing 5 % defibrinated horse blood (TCS Biosciences) at 42 °C in a modified atmosphere (85 % N2, 10 % CO2, and 5 % O2) generated with a VA500 workstation (Don Whitley Ltd.). Chloramphenicol was used at a concentration of 17 μg/ml for E. coli and 34 μg/ml for Campylobacter. Ampicillin was used at a concentration of 100 μg/ml.
Construction of C. jejuni expression vectors
The promoter regions of the C. jejuni NCTC 11168 porA, H. pullorum NCTC 12824 flaA and H. pylori 26695 ureI, genes were amplified with primer pairs porAXhoI-F/R, flaAXhoI-F/R and ureIXhoI-F/R respectively (Table 1), and cloned into the XhoI site immediately upstream of the cat cassette of pCJC1. A variant of the highly fluorescent gfp + gene [13] known as gfp TCD [14] was amplified using primer pair gfp-F/R (Table 1) and cloned into the NdeI site created in the promoter regions. The codon-optimised gfp Cj based on gfp TCD was synthesized (Eurofins) with flanking NdeI sites.
Transformation of C. jejuni
Electrocompetent C. jejuni cells were prepared and transformed with plasmid DNA using standard protocols [15].
GFP fluorescence monitoring of Campylobacter cultures
Cultures of C. jejuni were grown on blood agar for 24 h, resuspended in Heart Infusion (HI) broth and used to inoculate 2 ml HI broth supplemented with 5 % bovine foetal serum (BFS) to an OD600 of 0.05. Cultures were grown in a 6-well tissue culture dish in a modified atmosphere (as above) for approximately 16 h at 42 °C with shaking at 125 rpm. When cultures reached mid-log phase (between OD600 0.2 and 0.4) they were harvested by centrifugation, washed in PBS and resuspended to an OD600 of 1.0. A 10-fold dilution series of the bacterial suspension was made and 180 μl added in triplicate to a black-walled 96-well plate with transparent base. Both OD600 and fluorescence (excitation at 485/20 nm and emission at 528/20 nm) were measured on a Bio-Tek Synergy HT plate reader. Fluorescence in arbitrary units (AU) was calculated by dividing the Relative Fluorescence Units (RFU) by the corresponding OD600 value.
Immunodetection of recombinant proteins
C. jejuni strains expressing GFP were grown as for fluorescence measurement described above and whole cell lysates prepared after normalizing by OD600. Western blots were performed using mouse anti-GFP antibody (Sigma) and goat anti-mouse secondary antibody (Li-Cor).
Preparation of C. jejuni for fluorescence microscopy
C. jejuni strains expressing GFP were grown as above and approximately 108 cells were washed in 1 ml of phosphate buffered saline (PBS) and resuspended in 50 μl of PBS. Approximately 10 μl of this suspension was spread on a glass slide, dried, heat fixed and 50 μl of 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) added. Following incubation at room temperature for 20 min, slides were washed three times in PBS. Glass coverslips were adhered using Mowiol 4–88 and left to dry before imaging (see below).
HeLa cell infection by C. jejuni
HeLa cells cultured in Dulbecco’s Modified Eagles Medium (DMEM) were seeded into 6-well plates containing glass coverslips at 1.5 x 105 cells per well, and incubated for 24 h at 37 °C with 5 % CO2, to obtain a final density of 5 x 105 cells per well. Cells were washed with Dulbecco’s Phosphate Buffered Saline (PBSD) twice before infection. Mid-log cultures of C. jejuni grown in HI broth supplemented with 5 % BFS were harvested, washed twice in 1 ml PBS and added to HeLa cell cultures at a multiplicity of infection (MOI) of 10. Plates were incubated for a further 2 h, washed three times with PBSD and serum-free medium containing 50 μg/ml gentamicin added. After 1 h cover slips were removed, incubated in PBS containing 3 % paraformaldehyde for 20 min at room temperature and washed three times in PBS, the last wash containing 10 mM glycine (pH 8.5). Coverslips were then incubated in PBS containing 0.1 % Triton X-100 for 4 min at room temperature, washed three times in PBS, stained with DAPI as above and with the fluorescent actin stain phalloidin-Atto590 (Sigma) at 1:400 dilution followed by three final PBS washes.
Fluorescence microscopy of C. jejuni and HeLa cells
Images were collected on an Olympus BX51 upright microscope using a 40x/ 0.75 Plan Fln or 60x/ 0.65-1.25 Plan Fln objective and captured using a Coolsnap EZ camera (Photometrics) through MetaVue Software (Molecular Devices). Specific band pass filter sets for DAPI, FITC and Texas Red were used for visualizing DAPI, GFP and phalloidin-Atto590, respectively. Images were processed and analysed using ImageJ (http://rsb.info.nih.gov/ij).
Results
Design of novel C. jejuni chromosomal expression systems
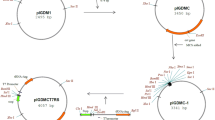
We have previously used a vector for genetic complementation in C. jejuni consisting of a 2179 bp fragment (corresponding to nt 205297–207475 inclusive of the C. jejuni NCTC 11168 genome sequence) cloned into pUC18 with a chloramphenicol resistance (cat) cassette cloned into a central SpeI site [11]. The cat gene is thus flanked by regions from pseudogene Cj0223, so that electroporation of this vector into C. jejuni cells results in integration, through a double recombination event, of the cat cassette and gene-of-interest onto the chromosome within Cj0223. This vector was modified by introduction of BglII/XhoI and NcoI/NheI sites flanking the cat cassette to produce plasmid pDENNIS [16]. This facilitated insertion of genes either upstream or downstream of cat and was used successfully for genetic complementation in C. jejuni [16]. We have now renamed pDENNIS as pCJC1 (Fig. 1). To further develop this vector providing increased and more flexible expression levels of genes introduced onto the C. jejuni chromosome, we have modified pCJC1 by cloning three distinct promoter regions, including native Shine-Dalgarno (SD) sequences and associated start codons (Fig. 2) immediately upstream of the cat gene (Fig. 1). Promoter regions (described below) were selected from Campylobacter and Helicobacter species based on previous evidence of their relatively high activity.

Construction of expression vectors for integration onto the C. jejuni chromosome. Construction of the pCJC series of plasmids for integration in the C. jejuni 11168 pseudogene Cj0223. A fragment of Cj0223 cloned into pUC18 is interrupted by a chloramphenicol resistance cassette (cat) at a unique SpeI site. Promoter regions with Shine-Dalgarno (SD) sites were cloned into the XhoI site followed by insertion of gfp at the NdeI site. Three promoter regions were used: H. pylori ureI to create pCJC2, H. pullorum flaA to create pCJC3 and C. jejuni porA to create pCJC4

Promoter regions employed in this study. The H. pylori 26695 ureI promoter region is previously characterised, the C. jejuni 11168 porA and H. pullorum NCTC 12824 flaA promoters are putative based on known consensus sequences. Boxed sequences denote known or putative −10 and −35 regions with the start codon of the associated open reading frames in bold capitals. Sequences were amplified by PCR to include 5′ and 3′ restriction sites and italicised bases were altered to “CAT” to create an NdeI site at the 3′ end
The first, (P porA ), is from the C. jejuni NCTC 11168 porA gene. Under several different growth conditions porA transcript is one of the most abundant in the transcriptome [17, 18]. Although transcription from P porA has been studied [19, 20], the promoter has not been mapped in detail. Our in silico analysis of the porA upstream region identified a putative SD site and a σ70-type promoter approximately 170 bp upstream of the start codon (Fig. 2). The second σ70-type promoter region chosen, P ureI from the H. pylori ureI gene (Fig. 2), was previously used in a plasmid-based inducible expression system for H. pylori [21]. The third promoter (P flaA ) is from the Helicobacter pullorum NCTC 12824 flaA gene (Fig. 2), a species studied in our laboratory. The flaA σ28-dependent promoters from C. jejuni [22] and H. pylori [23] are well characterized. A σ28 type promoter and SD site were identified upstream of H. pullorum flaA although the putative −10 and −35 regions are significantly further upstream (154 bp) compared to those from C. jejuni (54 bp) and H. pylori (57 bp) (Additional file 1: Figure S1).
Each promoter region with cognate SD site and ATG start site was PCR amplified to include 5′ and 3′ XhoI restriction sites allowing insertion into the unique XhoI site upstream of the pCJC1 cat cassette (Fig. 1). Primers were designed so that an NdeI site was placed adjacent to the XhoI site located immediately upstream of the cat cassette (Fig. 1). The NdeI recognition sequence of CA*TATG and primer design was such that the three bases preceding the start codon were altered to CAT to create the NdeI site (Fig. 2). This potentially allows introduction of a gene at the NdeI site creating a translational fusion to the start codon without altering the spacing between SD and start codon. Plasmids constructed in this way containing the P ureI , P flaA and P porA promoters were named pCJC2, pCJC3 and pCJC4 respectively (Fig. 1).
Analysis of relative expression levels in Campylobacter jejuni 11168
To test the relative expression levels from each of the promoter regions in C. jejuni we commercially synthesised a C. jejuni codon-optimised version of the gfp + gene [13, 23] and this was named gfp Cj (Additional file 2: Figure S2, GenBank accession KP994992). The gfp Cj gene was PCR amplified with 5′ and 3′ NdeI sites using primers gfp-F and gfp-R (Table 1) and inserted into pCJC2, pCJC3 and pCJC4 in the same transcriptional orientation as the cat gene, creating plasmids pCJC2gfpCj, pCJC3gfpCj and pCJC4gfp Cj respectively (Fig. 1). These C. jejuni suicide vectors were electroporated into strain 11168 and transformants identified in which a double recombination event had occurred at the Cj0223 locus. Strains constructed in this way were termed 11168gfp2, 11168gfp3 and 11168gfp4 and no significant effect on growth compared to the parental strain was observed (data not shown). To investigate promoter strength, relative fluorescence of early/mid-exponential phase cells of C. jejuni NCTC 11168 and the three gfp Cj expressing strains was measured in a 96-well plate reader (see Methods).
All three strains (11168gfp2, 11168gfp3 and 11168gfp4) displayed significantly higher fluorescence than wild-type 11168 cells. The levels of fluorescence from 11168gfp2, 11168gfp3 and 11168gfp4 were 106.0 (+/− 2.1 SE), 197.6 (+/− 23.1 SE) and 4002.9 (+/− 252.5 SE) arbitrary fluorescence units /OD600 respectively reflecting activity of their corresponding promoters P ureI , P flaA and P porA . Parallel Western blotting experiments of standardized whole-cell lysates from 11168gfp2, 11168gfp3 and 11168gfp4 using an anti-GFP antibody confirmed the gfp expression level pattern of P ureI < P flaA < P porA (Fig. 3). The varying expression levels of these three promoter regions will allow a choice of promoter for recombinant gene expression in C. jejuni based upon the desired application.

GFP levels from single copy chromosomal gfp Cj in C. jejuni strains 11168gfp2, 11168gfp3 and 11168gfp4. GFP detected by Western blotting in whole-cell lysates from strains 11168gfp2 (ureI), 11168gfp3 (flaA) and 11168gfp4 (porA)
Cultures of the C. jejuni 11168gfp4 strain with the PporA promoter driving expression of gfp Cj displayed very high fluorescence levels and so we tested its utility for in vitro imaging. Broth-grown cultures of 11168gfp4 were stained with DAPI and visualised for DAPI and GFP fluorescence by standard fluorescence microscopy (see Methods). Cells were clearly visible by GFP fluorescence (Fig. 4a) and fluorescence levels appeared consistent across individual cells as judged by comparison with DAPI staining, indicating homogenous levels of GFP production in the population, important for in vitro and in vivo experiments. We therefore used strain C. jejuni 11168gfp4 to infect HeLa cells in a basic cell invasion assay to determine if fluorescence levels were sufficient to monitor intracellular C. jejuni. Mid-log phase C. jejuni grown in broth were used to infect 24 h HeLa cells with an MOI of 10 prior to treatment with gentamicin to kill extracellular bacteria. Fixed HeLa cells were viewed by fluorescence microscopy following staining of cellular actin. Highly fluorescent intracellular C. jejuni cells were readily visualized within the actin and DAPI-stained HeLa cells (Fig. 4b). These observations confirm the utility of the combination of high activity promoter with insertion onto the C. jejuni chromosome to produce strains that stably express genes at high level. .

Fluorescent microscopy of C. jejuni 11168gfp4 cells. a. Fluorescent imaging of cells of wild-type C. jejuni NCTC 11168 and the 11168gfp4 strain expressing chromosomal gfp Cj driven by the C. jejuni promoter P porA . b. Fluorescent imaging of gentamicin-treated HeLa cells, 3 h post-infection with C. jejuni NCTC 11168gfp4 expressing chromosomal gfp Cj driven by the C. jejuni promoter P porA . Cells were stained with DAPI (blue) and phalloidin (red) with GFP fluorescence in green
Discussion
Genetic tools for the expression of recombinant genes are important for a number of applications and the lack of such tools for C. jejuni has been a limiting factor in experimental design. Here we have described a flexible set of constructs for the stable expression of genes at relatively low, medium and very high levels after chromosomal integration downstream of three distinct promoters. These expression levels can be used for applications such as functional complementation when low/medium expression levels might be desired and for the production and purification of specific proteins employing higher expression levels. The utility of very high-level expression from the P porA promoter was demonstrated by creating the highly fluorescent C. jejuni 11168gfp4 strain. The constructs described (pCJCgfp2, pCJCgfp3 and pCJCgfp4) could also be used to produce gfp translational fusions for determining the cellular localisation of specific proteins.
We have used a method for integrating genes onto the C. jejuni chromosome that targets pseudogene Cj0223 to minimize potential for a deleterious effect on cells. Cj0223 contains multiple frame-shift mutations/in-frame stop codons and is therefore non-functional and has been previously used to insert genes onto the C. jejuni chromosome [11]. The Cj0223 pseudogene is also present in most commonly used strains of C. jejuni so that this approach should be generally applicable.
The promoter elements used in this study were chosen based on previous studies describing either their activity or their utility in similar expression systems. The C. jejuni porA promoter is one of the strongest constitutive promoters in recent RNA-seq transcriptome studies, and PorA is the dominant protein in SDS-PAGE of C. jejuni whole-cell lysates [17, 18]. Promoter P flaA from H. pullorum was used as high levels of FlaA protein are produced in both C. jejuni and H. pylori. Promoter P ureI from H. pylori is a constitutive promoter successfully used in a H. pylori plasmid-based inducible expression system [21, 24]. To measure activity of these promoters we employed GFP as a reporter. Codon optimization of gfp for expression in C. jejuni more than doubled fluorescence observed with the P porA promoter (data not shown) highlighting the importance of considering specific translational features of host background. Indeed this gfp Cj allele might be considered for more general use in C. jejuni for applications such as a reporter gene for transcriptional and translational analysis and as a fusion partner for protein localization and quantitation. Comparison of fluorescence levels for strains expressing gfp from P ureI , P flaA and P porA demonstrated an approximately doubling of fluorescence with P flaA compared to P ureI and a further approximately twenty-fold increase with P porA . We propose that P porA should be the promoter of choice when high-level expression of recombinant proteins is desired although potential toxicity issues should be considered. The high level expression from P porA is also very useful for protein purification. High level expression of genes to produce specific proteins for structural/functional characterisation is generally performed in E. coli as this allows rapid and inexpensive production of large amounts of biomass and hence protein. However in certain circumstances where proteins may be post-translationally modified, for example by the protein glycosylation systems found in Campylobacters and some Helicobacter species, then production in these particular species is required to produce appropriately modified proteins. We successfully used promoter P porA to drive expression of hgpA encoding an N-linked glycoprotein in H. pullorum with sufficient yields for purification of milligrams of HgpA glycoprotein (data not shown).
In summary, the vectors presented in this work provide a useful set of tools to aid in molecular studies of this important bacterial pathogen and the principles involved in designing these tools should be generally applicable to other bacteria. Additionally we have constructed a new gfp allele that in combination with the expression systems described produced a highly fluorescent C. jejuni strain expressing gfp Cj from the chromosome for potential use in both in vitro and in vivo infection studies. The advent of synthetic biology and the affordable technology of gene synthesis will facilitate further development of these and other systems to produce a valuable genetic toolbox to aid in Campylobacter and Helicobacter research.
Conclusions
We have constructed vectors allowing recombination-mediated incorporation of genes onto the C. jejuni chromosome downstream of one of three promoters with varying expression levels. These vectors will be useful for genetic complementation, expression of genes at relatively high levels and construction of GFP translational fusions in this important bacterial pathogen.
Abbreviations
- cat :
-
Chloramphenicol resistance cassette
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DMEM:
-
Dulbecco’s Modified Eagles Medium
- GFP:
-
Green fluorescent protein
- LB:
-
Luria Bertani
- MOI:
-
Multiplicity of infection
- PBS:
-
Phosphate buffered saline
- PBSD:
-
Dulbecco’s phosphate buffered saline
References
Friedman CR, Niemann J, Wegener HC, Tauxe R. Epidemiology of Campylobacter jejuni infection in the United States and other industrialized nations. In: Nachamkin I, Blaser MJ, editors. Campylobacter. 2nd ed. Washington DC: ASM Press; 2000. p. 121–38.
Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Hoekstra RM. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis. 2011;17:7–15.
Havelaar AH, Ivarsson S, Lofdahl M, Nauta MJ. Estimating the true incidence of campylobacteriosis and salmonellosis in the European Union, 2009. Epidemiol Infect. 2012;141:293–302.
Labigne-Roussel A, Courcoux P, Tompkins L. Gene disruption and replacement as a feasible approach for mutagenesis of Campylobacter jejuni. J Bacteriol. 1987;178:1704–8.
Yao R, Alm RA, Trust TJ, Guerry P. Construction of new Campylobacter cloning vectors and a new mutational cassette. Gene. 1993;16:127–30.
Labigne-Roussel A, Harel J, Tompkins L. Gene transfer from Escherichia coli to Campylobacter species: development of shuttle vectors for genetic analysis of Campylobacter jejuni. J Bacteriol. 1988;169:5320–3.
Holt JP, Grant AJ, Coward C, Maskell DJ, Quinlan JJ. Identification of Cj1051c as a major determinant for the restriction barrier of Campylobacter jejuni strain NCTC11168. Appl Environ Microbiol. 2012;78(22):7841–8.
Karlyshev AV, Wren BW. Development and application of an insertional system for gene delivery and expression in Campylobacter jejuni. Appl Environ Microbiol. 2005;71:4004–13.
Gaskin DJH, van Vliet AHM, Pearson BM. The Campylobacter genetic toolbox: development of tractable and generally applicable genetic techniques for Campylobacter jejuni. Zoon Publ Health. 2007;54 Suppl 1:101.
Thomas MT, Shepherd M, Poole RK, van Vliet AH, Kelly DJ, Pearson BM. Two respiratory enzyme systems in Campylobacter jejuni NCTC 11168 contribute to growth on L-lactate. Environ Microbiol. 2011;13(1):48–61.
Hitchen P, Brzostek J, Panico M, Butler JA, Morris HR, Dell A, et al. Modification of the Campylobacter jejuni flagellin glycan by the product of the Cj1295 homopolymeric-tract-containing gene. Microbiology. 2010;156:1953–62.
Shaw FL, Mulholland F, Le Gall G, Porcelli I, Hart DJ, Pearson BM, et al. Selenium-dependent biogenesis of formate dehydrogenase in Campylobacter jejuni is controlled by the fdhTU accessory genes. J Bacteriol. 2012;194:3814–23.
Scholz O, Thiel A, Hillen W, Niederweis M. Quantitative analysis of gene expression with an improved green fluorescent protein. Eur J Biochem. 2000;267:1565–70.
Corcoran CP, Cameron AD, Dorman CJ. H-NS silences gfp, the green fluorescent protein gene: gfp TCD is a genetically remastered gfp gene with reduced susceptibility to H-NS-mediated transcription silencing and with enhanced translation. J Bacteriol. 2010;192:4790–3.
Van Vliet AHM, Wood AC, Henderson J, Wooldridge K, Ketley JM. Genetic manipulation of enteric Campylobacter species. Methods Microbiol. 1998;27:407–19.
Gundogdu O, Mills DC, Elmi A, Martin MJ, Wren BW, Dorrell N. The Campylobacter jejuni Transcriptional Regulator Cj1556 Plays a Role in the Oxidative and Aerobic Stress Response and Is Important for Bacterial Survival In vivo. J Bacteriol. 2011;193:4238–49.
Chaudhuri RR, Yu L, Kanji A, Perkins TT, Gardner PP, Choudhary J, et al. Quantitative RNA-seq analysis of the Campylobacter jejuni transcriptome. Microbiology. 2011;157:2922–32.
Butcher J, Stintzi A. The transcriptional landscape of Campylobacter jejuni under iron replete and iron limited growth conditions. PLoS One. 2013;8(11):e79475.
Dedieu L, Pagès JM, Bolla JM. Environmental regulation of Campylobacter jejuni major outer membrane protein porin expression in Escherichia coli monitored by using green fluorescent protein. Appl Environ Microbiol. 2002;68:4209–15.
Malik-Kale P, Parker CT, Konkel ME. Culture of Campylobacter jejuni with sodium deoxycholate induces virulence gene expression. J Bacteriol. 2008;190:2286–97.
Boneca IG, Ecobichon C, Chaput C, Mathieu A, Guadagnini S, Prévost MC, et al. Development of inducible systems to engineer conditional mutants of essential genes of Helicobacter pylori. Appl Environ Microbiol. 2008;74:2095–102.
Guerry P, Logan SM, Thornton S, Trust TJ. Genomic organization and expression of Campylobacter flagellin genes. J Bacteriol. 1990;172:1853–60.
Leying H, Suerbaum S, Geis R, Haas R. Cloning and genetic characterization of a Helicobacter pylori flagellin gene. Mol Microbiol. 1992;6:2863–674.
Akada JK, Shirai M, Takeuchi H, Tsuda M, Nakazawa T. Identification of the urease operon in Helicobacter pylori and its control by mRNA decay in response to pH. Mol Microbiol. 2000;36:1071–84.
Acknowledgements
This work was funded by the UK Biotechnology and Biological Science Research Council (BBSRC) grant BB/H017542/1. We acknowledge Dr Jiahui Wang, Dr Jane King and Miss Aini Zain at The University or Manchester for their help in microscopy sample preparation and HeLa cell infection. The fluorescence microscopy was performed in the Bioimaging Facility at the University of Manchester with microscopes purchased with grants from the BBSRC, the Wellcome Trust and the University of Manchester Strategic Fund. We thank Peter March and Roger Meadows for their technical help.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AJJ cloned promoters, constructed C. jejuni strains expressing gfp, measured expression levels, performed microscopy and drafted the manuscript. JAB produced plasmid pCJC1 and BWW directed the research. DL revised the manuscript and directed the research. All authors read and approved the manuscript.
Additional files
Additional file 1: Figure S1.
Sigma 28-dependent promoters of flaA genes from C. jejuni NCTC 11168, H. pylori 26695 and H. pullorum NCTC 12824. C. jejuni and H. pylori promoters are characterized [22, 23] and the putative promoter for H. pullorum is uncharacterized. Boxed sequences denote -10 and -35 regions, text in bold uppercase the flaA open reading frame start codon, text in bold the transcriptional start site and text in bold and italics the Shine-Dalgarno sequences. (DOCX 135 kb)
Additional file 2: Figure S2.
Nucleotide sequence of the C. jejuni codon-optimised gfp gene (gfp Cj). (DOCX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jervis, A.J., Butler, J.A., Wren, B.W. et al. Chromosomal integration vectors allowing flexible expression of foreign genes in Campylobacter jejuni . BMC Microbiol 15, 230 (2015). https://doi.org/10.1186/s12866-015-0559-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-015-0559-5