
Abstract
Background
The black-lip rock oyster (Saccostrea echinata) has considerable potential for aquaculture throughout the tropics. Previous attempts to farm S. echinata failed due to an insufficient supply of wild spat; however, the prospect of hatchery-based aquaculture has stimulated renewed interest, and small-scale farming is underway across northern Australia and in New Caledonia. The absence of knowledge surrounding the population genetic structure of this species has raised concerns about the genetic impacts of this emerging aquaculture industry. This study is the first to examine population genetics of S. echinata and employs both mitochondrial cytochrome c oxidase subunit I gene (COI) and single nucleotide polymorphism (SNP) markers.
Results
The mitochondrial COI data set included 273 sequences of 594 base pair length, which comprised 74 haplotypes. The SNP data set included 27,887 filtered SNPs for 272 oysters and of these 31 SNPs were identified as candidate adaptive loci. Data from the mitochondrial COI analyses, supports a broad tropical Indo-Pacific distribution of S. echinata, and showed high haplotype and nucleotide diversities (0.887–1.000 and 0.005–0.008, respectively). Mitochondrial COI analyses also revealed a ‘star-like’ haplotype network, and significant and negative neutrality tests (Tajima’s D = − 2.030, Fu’s Fs = − 25.638, P < 0.001) support a recent population expansion after a bottleneck. The SNP analyses showed significant levels of population subdivision and four genetic clusters were identified: (1) the Noumea (New Caledonia) sample location; (2) the Bowen (north Queensland, Australia) sample location, and remaining sample locations in the Northern Territory, Australia (n = 8) were differentiated into two genetic clusters. These occurred at either side of the Wessel Islands and were termed (3) ‘west’ and (4) ‘east’ clusters, and two migrant individuals were detected between them. The SNP data showed a significant positive correlation between genetic and geographic distance (Mantel test, P < 0.001, R2 = 0.798) and supported isolation by distance. Three candidate adaptive SNPs were identified as occurring within known genes and gene ontology was well described for the sex peptide receptor gene.
Conclusions
Data supports the existence of genetically distinct populations of S. echinata, suggesting that management of wild and farmed stocks should be based upon multiple management units. This research has made information on population genetic structure and connectivity available for a new aquaculture species.
Similar content being viewed by others


Background
For farmed marine species, understanding population structure and how populations respond to the environment can inform broodstock selection, translocation boundaries, and aquaculture induced evolution [1, 2]. Potential impacts from aquaculture relate to the introgression of exotic alleles, caused by processes such as: interbreeding or outbreeding of wild and cultivated populations, the outcompeting of native alleles by introduced alleles, and translocations, all of which can negatively alter the genetic diversity of remnant populations [3]. Therefore, an understanding of natural population genetics, before aquaculture occurs, is necessary for the proactive management of these impacts [4, 5]. Understanding the genetics of natural populations also provides potential benefits for improving aquaculture and biosecurity practices as many adaptive traits in the wild are important production traits, such as; growth rate, environmental tolerances, and disease resistance [6, 7].
Many methods exist to investigate population genetics, and those such as mitochondrial genes provide useful tools for identifying and improving our understanding of relationships among species [8, 9]. More sophisticated and increasingly affordable methods are becoming available for application on non-model species; which often lack genomic resources and includes many species used for aquaculture [10]. The advent of next generation sequencing technologies and restriction-site associated DNA (RADseq) genotyping methods, has enabled delivery of genome-wide single nucleotide polymorphisms (SNPs) for any organism (e.g. non-model species), at reasonable costs [11, 12]. Genome-wide SNPs can reveal fine-scale patterns of population structure, dispersal capabilities, population assignment, and detect signatures of selection [13,14,15], with much higher resolution than traditional markers (e.g. allozymes and microsatellites). The use of these technologies in the detection of fine-scale structure and signatures of selection are important for determining management units, particularly in populations that may otherwise appear homogenous in their distribution [10].
Marine bivalves are characterised by large population sizes, high fecundity, and planktonic larvae with considerable dispersal potential [14, 16, 17]. In sessile bivalve populations, such as oysters, gene flow is achieved through the dispersal of gametes which frequently results in high genetic diversity and weak genetic differentiation [18]. This is true for the tropical oyster Crassostrea corteziensis in the Gulf of California where allozyme analyses revealed a single panmictic population, possessing high genetic diversity [19]. However, deviations from this trend can occur, usually as a result of larval behaviour, environmental gradients, anthropogenic effects, and physical barriers [20,21,22]. For example, analyses of mitochondrial and SNP data revealed many subpopulations of Crassostrea virginica throughout the Gulf of Mexico, despite its potential for high gene flow [23]. Similar results of population differentiation in a species with high dispersal capabilities, have been reported for Crassostrea iredalei throughout Malaysia [24] and Ostrea edulis along the European coast [25]. Therefore, without detailed population genetic analyses, predictions cannot be made regarding the extent of population genetic structure of oyster species.
The black-lip rock oyster, Saccostrea echinata (Quoy and Gaimard, 1835), is a tropical marine bivalve that is reported to have broad geographic distribution throughout the Indo-Pacific [26]. Research is currently underway to improve the commercial aquaculture potential of S. echinata due to the success of small-scale farms (< 1 t per annum) in northern Australia, eastern Australia, and New Caledonia (JR Collison pers. comm. 2018). Production based on wild seed supply has been a bottleneck for the industry [27], and this is still the case. Development of hatchery protocols for this species is therefore considered a prerequisite for advancement and progress to commercial scale hatchery production is being made [28, 29]. Larvae of S. echinata are planktonic and development occurs over approximately 21 days in the hatchery, after which metamorphosis and settlement takes place [29]. Larvae may conceivably travel hundreds of kilometres before settlement, which leads to the hypothesis that wild populations possess low levels of genetic structure [30]. However, studies on related oyster species have reported both panmictic [19, 30] and highly divergent [23, 24] population structures. To achieve the genetic conservation of oyster stocks in the face of developing aquaculture efforts, a sound knowledge of population genetic structure is essential. A genetic stock assessment of natural S. echinata populations, before commencement of commercial-scale aquaculture, would provide biological information upon which responsible management and hatchery production could be based. This study aims to fill this knowledge gap by analysing both mitochondrial and nuclear DNA markers to assess the population genetic structure and the neutral and adaptive genetic diversity of S. echinata across northern Australia.
Results
Mitochondrial COI data
The mitochondrial cytochrome c oxidase subunit I gene (COI) data set comprised 273 sequences of 594 base pair (bp) length, which contained 74 different haplotypes. Haplotype and nucleotide diversities were high (0.887–1.000 and 0.005–0.008, respectively) and similar among all sample locations except Bowen, which were markedly lower (0.571 and 0.001, respectively) (Fig. 1; Table 1). Anuru Bay had the highest haplotype diversity (1.000 ± 0.022); every individual had a different haplotype (Table 1). Noumea had the highest number of private haplotypes (9), while Bukudal had the lowest (zero). No haplotypes were shared among all locations (Table 1). Kimura 2-parameter (K2P) distance between all localities in this study and from those obtained from GenBank (i.e. Malaysia, Taiwan, Japan and Western Australia) are provided in Additional file 1.

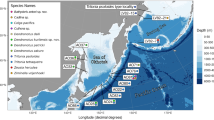
Map showing locations from which oysters (Saccostrea echinata) were sampled (circle markers). SMP, Semporna (4°29′6″N 118°36′12″E); TWN, Taiwan (22°45′24″N 121°09′02″E); TKT, Taketomi Island (24°21′36″N 123°44′60″E); CNB, Cone Bay (16°30′19″S 123°34′55″E); PRL, Pirlangimpi (11°15′07″S 130°20′53″E); MLK, Milikapiti (11°22′5″S 130°34′21″E); ANR, Anuru Bay (11°43′23″S 133°20′35″E); WGU, Wigu (11°38′49″S 133°25′23″E); MRN, Mooroongga Island (11°56′06″S 135°02′47″E); NHL, Nhulunbuy (12°14′14″S 136°40′30″E); BKD, Bukudal (13°00′40″S 136°37′41″E); UMB, Umbakumba (13°39′03″S 136°40′45″E); BWN, Bowen (19°56′54″S 148°08′55″E); NUM, Noumea (22°17′21″S, 166°26′36″E)
Pairwise ϕST estimates of haplotype frequency between localities showed Bowen and Noumea (except for Noumea and Anuru Bay) as significantly different from all other populations (Table 2). Bukudal was also significantly different from both Pirlangimpi and Wigu, and Nhulunbuy was significantly different from Pirlangimpi. No other localities were different from one another. However, the level of genetic differentiation did not depend on the geographic distance separating sample locations (Mantel test, P = 0.104, R2 = 0.520). Likewise, the haplotype network indicates lack of population genetic structure between populations and does not support geographical haplotype groups (Fig. 2). The haplotype network showed two larger ‘ancestral’ haplotypes with many (28 and 7) unique haplotypes radiating from them (Fig. 2). Analyses of all locations together resulted in negative mean Tajima’s D and Fu’s Fs that were significant (Table 2). Negative Tajima’s D indicates an excess of low frequency polymorphisms and negative Fu’s Fs indicates an excess of alleles expected from recent directional selection or population growth [24, 31]. Phylogenetic analysis with ostreid COI sequences placed all S. echinata samples obtained in this study within ‘Lineage J’ as nominated by Sekino and Yamashita [32] (Additional file 2).

Median joining haplotype network of Saccostrea echinata mitochondrial COI sequence data. Each bar on the branch corresponds to a single nucleotide substitution. Larger circles contain specific frequency size
SNP data
Genotyping
Approximately 761 million sequence reads of 150 bp each were obtained, filtered and trimmed from the eight lanes of Illumina NextSeq500 sequencing. From these, approximately 15 million double-digest restriction site-associated DNA (ddRAD) loci from 282 individuals were identified. The data set was then filtered to 27,887 SNPs from 272 individuals which were used for further analyses (Table 3). BAYESCAN 2.1 and PCAdapt detected 51 and 483 outliers, respectively. Of these, 31 loci were detected by both methods and considered as candidate adaptive loci (Table 3). Subsequent analyses were conducted for the putatively neutral data set of 27,856 loci and the candidate adaptive data set of 31 loci.
Genetic diversity and population structure
The number of alleles and the allelic richness were similar among localities; the number of alleles ranged from 32,023–45,861 (neutral) and 37–55 (candidate adaptive), and the allelic richness ranged from 1.083–1.444 (neutral) and 1.130–1.851 (candidate adaptive). Bowen had the lowest number of alleles, with 27,585 (neutral) and 28 (candidate adaptive), and an allelic richness of 0.972 (neutral) and 0.892 (candidate adaptive) (Table 4). Observed heterozygosity was lower than expected heterozygosity for all populations (Table 4). Mean expected and observed heterozygosity were higher for the candidate adaptive SNPs than the neutral SNPs and the inbreeding coefficient was similar and positive among localities (Table 4). Estimates of effective population size ranged from 212.8–infinity (neutral) and varied among sampling locations (Table 4). Infinite effective population size estimates and confidence intervals may be due to larger than expected sampling error [33]. Levels of population differentiation across localities were high with 39 out of 45 pairwise comparisons significantly different than zero (Table 5). Pairwise comparisons for the neutral data set ranged from zero (Milikapiti vs Pirlangimpi, Umbakumba vs. Nhulunbuy, and Bukudal vs. Nhulunbuy) to 0.058 (Noumea vs. Anuru Bay and Noumea vs. Wigu). Pairwise estimates that were not significant occurred between some of the Northern Territory populations, indicating greater homogeneity between these populations.
While both neutral and candidate adaptive SNPs detected population structure between sampling locations, the neutral loci detected greater differentiation (Fig. 3a–f). Discriminant analysis of principal components (DAPC) of all 10 sampling locations using all SNPs detected four clusters, with Noumea and Bowen forming their own clusters and the remaining localities from the Northern Territory, Australia separating into two clusters (Fig 3a). Analyses of only the eight Northern Territory sampling locations showed some mixing between the two clusters by the Mooroongga population (Fig. 3b). This pattern remained when analysing neutral and candidate adaptive SNPs separately (Fig. 3d,f). There was a significant positive correlation between genetic differentiation and the geographic distance separating sample locations (Mantel test, P < 0.001, R2 = 0.798).

Discriminant analyses of principal components (DAPC) of Saccostrea echinata samples by sampling locality. (a) For all 27,887 SNPs and all sample locations (b) For all 27,887 SNPs and only Northern Territory sample locations (c) For all 27,856 putatively neutral SNPs and all sample locations (d) For all 27,856 putatively neutral SNPs and only Northern Territory sample locations (e) For all 31 candidate adaptive SNPs and all sample locations (f) For all 31 candidate adaptive SNPs and only Northern Territory sample locations. PRL, Pirlangimpi; MLK, Milikapiti; ANR, Anuru Bay; WGU, Wigu; MRN, Mooroongga Island; NHL, Nhulunbuy; BKD, Bukudal; UMB, Umbakumba; BWN, Bowen; NUM, Noumea
Individuals were assigned to their region of origin with very high success; 98.6 and 100% of individuals were correctly assigned to population of origin, with both 10,000 SNP data sets. Two migrant individuals were detected and both were from the west population, specifically the Mooroongga sample location.
Functional annotation
The alignment of the 31 candidate sequences to the complete black-lip rock oyster transcriptome resulted in 19 hits. Three of these were successfully identified as belonging to known genes and are presented in Table 6: (1) SNP 2350 is situated in the gene Kmt5a, which is required for cell proliferation, probably by contributing to the maintenance of higher-order structure of DNA during mitosis; (2) SNP 18035 is situated in the gene FAXC that may play a role in neural, axonal development; and (3) SNP 34199 which is situated in the sex peptide receptor (SPR) gene. The SPR gene had the most well-described gene ontology annotation terms and has been reported to be involved in controlling reproductive and sleep behaviours in fruit flies (Drosophila melanogaster) [34, 35].
Discussion
This is the first study to investigate the population genetics of S. echinata and it utilises both mitochondrial and nuclear DNA markers. Results support the existence of genetically distinct populations of S. echinata, and this has implications for the management of wild and farmed stocks of this new aquaculture species, that should be considered as multiple management units.
Results from the mitochondrial COI analyses, are consistent with those of McDougall [36], and the observational data of Thomson [26], in that they support a broad tropical Indo-Pacific distribution of S. echinata; from at least Taketomi Island in Japan, to Noumea in New Caledonia, and across northern Australia; from Cone Bay in Western Australia, to Bowen in Queensland. Few studies have employed genetic data to assist in investigating the distribution of tropical oyster species within the Indo-Pacific region. The tropical rock oyster, C. iredalei, has been confirmed to occur throughout Malaysia, based on mitochondrial COI analyses [24]. Similar research has also been conducted with the tropical black-lip pearl oyster, Pinctada margaritifera, which confirmed broad Indo-Pacific distribution, based on SNP analyses [14]. Genetic confirmation of the geographic range of S. echinata is important for countries interested in aquaculture of this species, particularly those interested in hatchery production of spat, because culture conditions and techniques are species-specific [37, 38]. A recent review of the diversity and evolution of oysters (both temperate and tropical species) with a focus on molecular studies by Guo et al. [39] highlighted that balancing selection is a major force in shaping genetic variation among oyster populations. Strong balancing selection results from; different life stages (planktonic larvae and sessile adults), long-distance dispersal potential, and fluctuating environmental conditions, and results in high levels of genetic diversity [40, 41]. High genetic diversity has been shown across oyster populations that are panmictic, such as Crassostrea gigas in Japan [42], and highly divergent, such as C. virginica in America [43]. In the current study, analyses of mitochondrial COI data showed high haplotype and nucleotide diversities (excluding the Bowen location), that are similar to those reported for other molluscs, such as the oyster C. iredalei [24], the clam Anomalocardia brasiliana [44], and the Hawaiian limpet Cellana talcosa [45]. These studies [44, 45] have been compared in a summary of published haplotype and nucleotide diversity estimates by Goodall-Copestake et al. [46], to provide a standardised reference for comparison and improve the consistency of reporting for diversity estimates. High diversity estimates in wild populations, as reported in the current study, are a positive attribute for hatchery-based aquaculture and support the development of selective breeding programs [6, 10].
Relatively low haplotype and nucleotide diversity indices detected in Bowen samples may be due to unintentional sampling bias. These individuals were collected from an oyster farm and it is possible that they may have been collected from a single or few cohort(s). It is also probable that the Bowen population is close to the edge of the species range and many taxa show a general decline in genetic diversity towards range margins; a pattern known as the core-periphery hypotheses [14, 47, 48]. Despite Bowen and Noumea being the most distant sample locations, mitochondrial COI analyses did not support isolation by distance, and the haplotype network for S. echinata in this study did not support geographical haplotype groups. The ‘star-like’ haplotype network and congruence between Tajima’s D and Fu’s Fs overall negative and significant P-values suggest a recent population expansion after a bottleneck [24, 49].
In contrast to the lack of structure between sampled populations of S. echinata shown by mitochondrial COI analyses, the SNP analyses showed high levels of population subdivision. Greater differentiation of nuclear SNP markers, compared with mitochondrial COI markers, was similarly reported by Varney et al. [50] in a population genetic investigation of the eastern oyster, C. virginica, in the Gulf of Mexico. Mitochondrial DNA analysis is a useful first-pass marker for investigating genetic structure, however, it is not a suitable marker for the detection of fine spatial genetic structure [51], and it is therefore important to follow-up with higher resolution genomic markers [52], such as genome-wide SNPs [53,54,55].
In the current study, the SNP analyses clearly showed that the S. echinata populations sampled do not belong to a single panmictic unit. At a broad scale, S. echinata populations in Bowen and Noumea displayed substantial and significant divergence from all other populations. Within the Northern Territory, Australia, low but significant differentiation was observed among some of the sample locations and DAPC analyses identified two genetic clusters in this region (here termed ‘west’ and ‘east’). Some mixing was detected, by the DAPC analyses, between the west and east clusters in the Northern Territory, which was further confirmed by the detection of two migrant individuals at Mooroongga, which is part of the west cluster, that were genetically most similar to the Nhulunbuy sample location, which is part of the east cluster. Both the structure shown in the DAPC analyses and a significant positive correlation between genetic and geographic distance support isolation by distance occurring in sampled S. echinata populations.
Distinct genetic clusters within the Northern Territory suggest that larval dispersal is limited to within these two regions. These two clusters occur either side of the Wessel Islands, a chain of small islands extending 120 km northeast from the Napier Peninsula (Fig. 4), that are a potential physical barrier to S. echinata larval dispersal and thus gene flow. Furthermore, this island chain would have been a land barrier around seven thousand years ago, when sea levels were lower, which may be reflected in the genetic clusters identified [58]. In a similar study investigating population genetics of the sea cucumber, Holothuria scabra, in northern Australia, Gardner et al. [59] reported the same genetic clusters either side of the Wessel Islands. They suggested a hydrological barrier exists, due to prevailing surface currents (Fig. 4); currents on the west side carry larvae westward and on the ‘east’ side a circular current in the Gulf may entrain larvae, restricting the passive dispersal of H. scabra larvae, which have a similar larval duration to larvae of S. echinata [29, 59]. Gardner et al. [59] also proposed that the Arafura Basin, situated west of the Wessel Islands (Fig. 4), may act as a benthic barrier to larval dispersal, because rivers that drain into the Arafura Basin create an environment of low salinity [60]. The congruence between the findings of Gardner et al. [59] and the current study, support the need for further investigation to determine mechanisms underlying shared genetic patterns; such investigations may include a multi-species phylogeographic study of the region [52] and more sampling of S. echinata throughout the Gulf of Carpentaria.

Map showing locations from which oysters (Saccostrea echinata) were sampled within the Northern Territory, Australia. Different coloured circle markers indicate two genetic clusters (1) the west cluster (blue circle markers); PRL, Pirlangimpi (11°15′07″S 130°20′53″E); MLK, Milikapiti (11°22′5″S 130°34′21″E); ANR, Anuru Bay (11°43′23″S 133°20′35″E); WGU, Wigu (11°38′49″S 133°25′23″E); and MRN, Mooroongga Island (11°56′06″S 135°02′47″E); and (2) the east cluster (orange circle markers); NHL, Nhulunbuy (12°14′14″S 136°40′30″E); BKD, Bukudal (13°00′40″S 136°37′41″E); and UMB, Umbakumba (13°39′03″S 136°40′45″E). The dominant near-surface current circulation patterns and location of the Arafura Basin (indicated by the lined polygon) are illustrated (adapted from Schiller [56] and Geoscience Australia [57])
When undertaking population genetic analyses with SNPs it is important to consider both neutral markers, which provide useful information about neutral evolutionary processes, and candidate adaptive markers which are more likely to display signatures of selection [54]. In the current study neutral and candidate adaptive SNPs were analysed separately and together (when appropriate), and no major conflicts between resulting data sets were detected. One advantage for determining candidate adaptive SNPs is that they can identify genes under selection, and if these genes are known, then gene ontology may reveal locally important adaptations [53, 55]. In the current study of S. echinata, three candidate adaptive SNPs were successfully identified as belonging to known genes, and gene ontology was well described for the SPR gene. The SPR gene has been studied in fruit flies (D. melanogaster) and has been shown to be important for determining reproductive behaviour [34, 61, 62] and sleep stabilisation [35]. A molecular function of this gene is G protein-coupled peptide receptor activity which can be important in immunity, protein activation, and other molecular functions [34]. The SPR gene may play a role in adaptive differences between the S. echinata populations studied, such as differences in reproductive behaviours (e.g. the timing of reproductive cycles or of spawning). Further research is required to better understand the role of the SPR gene in relation to seascape genomics of S. echinata.
Management implications
Genetic information is an important part of fisheries and aquaculture management policy and has been used to identify management units for many commercially important species, including green-lip abalone (Haliotis laevigata) [2, 63], mangrove oysters (Crassostrea spp.) [64], Atlantic cod (Gadus morhua) [53], and black-lip pearl oysters (Pinctada margaritifera) [14]. Given this study’s findings of significant population differentiation and the presence of signatures of selection, the data supports the existence of genetically distinct populations of S. echinata. This suggests that management of wild and farmed S. echinata stocks should be based upon multiple management units, and new areas interested in farming this species should undergo genetic stock assessments to determine optimal management practises. Within the Northern Territory, Australia where most of the sample locations in this study occur, and where S. echinata aquaculture development is focused, two genetically distinct clusters exist, which may be differentially adapted to local environmental conditions. It is recommended that these distinct clusters be considered as management units; when broodstock are being selected for hatchery production, for the translocation of wild or hatchery-produced spat, and for any translocations of farmed stocks.
Conclusions
Use of both mitochondrial and nuclear DNA markers to assess the population genetics of S. echinata across northern Australia has made data on population genetic structure and connectivity available. This study confirmed that despite the potential for high levels of gene flow, S. echinata do not belong to a single panmictic unit. The development of management policy for culture practices and translocation of farmed oysters will become increasingly important as hatchery production develops and aquaculture operations expand. Furthermore, this study has provided high quality genome-wide SNP marker sets that can be utilised for future genetic research, such as the use of genetics in stock improvements (e.g. selective breeding programs), and to improve understanding of locally important adaptations in wild oyster populations.
Methods
Sample collection and genomic DNA extraction
In total, 273 oysters were sampled across 10 sampling sites; nine in Australia (from the Northern Territory and Queensland) and one in New Caledonia (Fig. 1). The sites chosen within Australia were those under consideration for commercial oyster farming. The New Caledonian sample collection was opportunistic, however, the site is also under consideration for farming. Between 15 and 37 oysters were collected randomly at each site, during low tide, from intertidal wild stocks before being transported live in a damp hessian bag, inside a foam box to the Northern Territory Government, Department of Primary Industry and Resources, Darwin Aquaculture Centre. A 0.5 cm2 of adductor muscle was excised from live oysters and stored in 96% ethanol in an Eppendorf tube. Exceptions to this method include samples collected from Bowen, Australia (BWN, Fig. 1) where oysters were collected from farmed stocks, and samples collected from Noumea, New Caledonia (NUM, Fig. 1) where processing of samples occurred in country. Genomic DNA (gDNA) was extracted using the DNeasy Blood and Tissue kit (QIAGEN®, Germany) following the manufacturer’s instructions with a final elution of 100 μL.
Analyses of mitochondrial COI data
Molecular techniques
A 594 bp fragment of the mitochondrial COI gene was amplified using the polymerase chain reaction (PCR) protocol. The ‘Folmer’ primers [9] were used and reaction conditions were: 1 cycle of 94 °C for 1 min, followed by 5 cycles of 94 °C for 40 s, 45 °C for 40 s, 72 °C for 1 min, followed by 35 cycles of 94 °C for 40 s, 51 °C for 40 s, 72 °C for 1 min and concluding with a final extension of 72 °C for 5 min. Integrity of gDNA was determined through gel electrophoresis.
Data analyses
Sequences were aligned using Geneious Alignment implemented within Geneious 11.1.5 (http://www.geneious.com/) and trimmed (sequence accession numbers provided in Additional file 3). Sequences were grouped into source localities for data analyses according to their geographical proximity (Fig. 1). Haplotype (h) and nucleotide (π) diversities were calculated using DnaSP 6.12.01 [65]. Tajima’s D and Fu’s Fs neutrality tests were performed in the program ARLEQUIN 3.5.2.2 [31, 66, 67]. Pairwise ϕST estimates of haplotype frequency between localities were also calculated using ARLEQUIN 3.5.2.2. To determine whether an isolation by distance pattern was prevalent in S. echinata, a genetic distance (ϕST) matrix was compared with a geographic distance (km) matrix using the R package ade4 [68]. Geographical distances were estimated as the shortest distance by sea between the locality pairs as determined by Google Earth. A median joining haplotype network was generated from the mitochondrial COI sequence data using PopART [69, 70] with the default settings. Sequences of S. echinata from previous studies (n = 21) [32, 71] were obtained from GenBank and also included in the network analyses. Sampling sites of these sequences include Semporna in Malaysia, Taiwan, and Taketomi Island in Japan (coordinates listed in Fig. 1). Sequences of S. echinata were also obtained from the Government of Western Australia (n = 4) from the sampling site Cone Bay (coordinates listed in Fig. 1). Evolutionary distances were estimated using the program MEGA-X with the K2P model [72]. Mitochondrial COI sequences were aligned with other partial ostreid COI sequences obtained from McDougall [35] and phylogenetic analysis was performed in Geneious 11.1.5 using the Tamura-Nei genetic distance model with the Neighbour-Joining tree building method and 100 nonparametric bootstrap replicates. Phylogenetic trees were visualised using FigTree [73].
Analyses of SNP data
Molecular techniques
Double-digest restriction site-associated DNA (ddRAD) libraries were constructed following a protocol modified from Peterson et al. [74]. Briefly, 200 ng of gDNA was digested using the restriction enzymes EcoRI and NlaIII. One of 48 unique bp barcodes was ligated to each individual library. Size selection of pooled digested-ligated fragments was conducted using Blue Pippin (Sage Science, USA). Libraries were amplified using PCR and single-read, 150 bp target length sequencing was conducted on an Illumina NextSeq500 platform with 150 cycles in high-output mode at the Australia Genome Research Facility (Melbourne, Australia).
Bioinformatics and genotyping
Libraries were demultiplexed using the process_radtags program in STACKS 1.47 [75]. Reads were allowed a maximum of two nucleotide mismatches (n = 2) and a minimum stack depth of three (m = 3) (ustacks module in STACKS, with default parameters). Then stacks were aligned de novo with each other to create a catalogue of putative RAD tags (cstacks module in STACKS, with a maximum of one nucleotide mismatch allowed (n = 1)). In the population module of STACKS, and following consecutive filtering steps, SNPs were retained in at least 70% of the individuals and 50% of the locations, had a minor allele frequency of at least 10%, and heterozygosity < 0.5. Analyses were restricted to one random SNP per locus (using the --write_random_snp option in the populations module). SNPs with more than 70% of missing data and individuals with more than 50% missing data were also eliminated using VCFtools [76]. Details of the number of SNPs kept after each filtering step are provided in Table 3. The resulting filtered VCF file (Additional file 4) was converted into the file formats necessary for the following analyses using PGDSpider 2.1.1.5 [77].
Data analyses
To identify SNPs potentially under neutral and adaptive selection a combination of two outlier detection approaches were used; BAYESCAN 2.1 and PCAdapt R package [78]. Outlier loci detected by both BAYESCAN and PCAdapt were conservatively selected as candidate adaptive loci (Additional file 5) and subsequent analyses were run for the neutral and outlier datasets separately. The level of observed heterozygosity (HO) and expected heterozygosity (HE) in each locality, as well as F statistics were calculated using GenoDive 2.0 [79]. The number of alleles (A) and allelic richness (AR) were calculated with the R packages PopGenReport and hierfstat, respectively [80]. Effective population size (NE) and 95% confidence intervals were estimated using the software NeEstimator v.2.1 using the linkage disequilibrium method [33] for neutral SNPs only as the method assumes neutrality. Population structure was investigated through DAPC analyses. DAPC was performed, and results were plotted using the R package adegenet [81]. To determine whether an isolation by distance pattern is prevalent in S. echinata, a genetic distance (FST) matrix was compared with a geographic distance (km) matrix using the R package ade4 [68]. Geographical distances were estimated as the shortest distance by sea between the locality pairs as determined by Google Earth.
Assignment tests were performed in GenoDive 2.0 [79] on two data sets of 10,000 SNPs that were randomly generated from the neutral SNP data set. Individuals from Bowen and Noumea were removed and the remaining individuals from the sample locations in northern Australia were merged to form two ‘populations’ (determined by the DAPC clusters). The west population included: Pirlangimpi, Milikapiti, Anuru Bay, Wigu, and Mooroongga Island. The east population included: Nhulunbuy, Bukudal, and Umbakumba. This was done to increase the accuracy of the assignment test, as they are moderately accurate (~ 78%) for populations with low levels of genetic subdivision (FST ~ 0.04) [82]. Pairwise FST values between the two population clusters was 0.03 for both data sets of 10,000 randomly chosen SNPs. In a given genotype, when the observed frequency of any allele was zero (a missing allele), the frequency of this allele was replaced by a fixed value of 0.005 as recommended by Paetkau et al. [83], to avoid the calculation of a multilocus likelihood of zero. A null distribution of likelihood values was generated using a Monte Carlo chain [84] for 5000 permutations. The Cornuet et al. [84] algorithm, with a statistical threshold calculated separately for every population based on an alpha-value of 0.05, was used [82]. Individuals with likelihood values originating from their sampling location (LH) inferior to this threshold are thus defined as putative migrants.
Functional annotation
The basic local alignment search tool (BLAST) function, incorporated in Geneious 11.1.5, was used to BLAST the 31 candidate adaptive SNPs against the complete, annotated transcriptome of the black-lip rock oyster, referred to as Saccostrea lineage J by McDougall [36]. A minimal E-value threshold of 1 × 10− 6 and the highest ‘Grade’ (a weighted score for the hit comprised of the E-value, the pairwise identity, and the coverage) for each hit were applied to the analyses. This yielded three candidate SNPs successfully identified as belonging to known genes, contained within the UniProtKB/ Swiss-Prot database [85]. Gene ontology annotation terms were then associated with the candidate SNPs.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its Additional files.
Abbreviations
- BLAST:
-
basic local alignment search tool
- Bp:
-
Base pair
- DAPC:
-
Discriminant analysis of principal components
- ddRAD:
-
Double-digest restriction site-associated DNA
- DNA:
-
Deoxyribonucleic acid
- gDNA:
-
Genomic deoxyribonucleic acid
- GO:
-
Gene ontology
- mitochondrial COI:
-
Mitochondrial cytochrome c oxidase subunit I gene
- PCR:
-
Polymerase chain reaction
- RADseq:
-
Restriction-site associated DNA
- SNP:
-
Single nucleotide polymorphism
- SPR gene:
-
Sex peptide receptor gene
References
Besnier F, Glover KA, Lien S, Kent M, Hansen MM, Shen X, et al. Identification of quantitative genetic components of fitness variation in farmed, hybrid and native salmon in the wild. Heredity. 2015;115(1):47–55.
Sandoval-Castillo J, Robinson NA, Hart AM, Strain LWS, Beheregaray LB. Seascape genomics reveals adaptive divergence in a connected and commercially important mollusc, the greenlip abalone (Haliotis laevigata), along a longitudinal environmental gradient. Mol Ecol. 2017;27(7):1603–20.
Bert TM, Nielsen JL. Ecological and genetic implications of aquaculture activities. Dordrecht: Springer; 2007.
Palsboll PJ, Berube M, Allendorf FW. Identification of management units using population genetic data. Trends Ecol Evol. 2007;22:11–6.
Frankham R. Challenges and opportunities of genetic approaches to biological conservation. Biol Conserv. 2010;143:1919–27.
Gjedrem T. Genetic improvement for the development of efficient global aquaculture: a personal opinion review. Aquaculture. 2012;344–349:12–22.
Bernatchez L, Wellenreuther M, Araneda C, Ashton DT, Barth JMI, Beacham TD, et al. Harnessing the power of genomics to secure the future of seafood. Trends Ecol Evol. 2017;32(9):665–80.
Brown WM. The mitochondrial genome of animals. In: McIntyre RJ, editor. Molecular evolutionary genetics. New York: Plenum Press; 1985. p. 95–130.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome coxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3(5):294–9.
McAndrew B, Napier J. Application of genetics and genomics to aquaculture development: current and future directions. J Agr Sci. 2010;149:143–51.
Davey JW, Blaxter ML. RADSeq: next-generation population genetics. Brief Funct Genomics. 2010;9(5–6):416–23.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12(7):499–510.
Limborg MT, Helyar SJ, Bruyn MD, Taylor MI, Nielsen EE, Ogden R, et al. Environmental selection on transcriptome-derived SNPs in a high gene flow marine fish, the Atlantic herring (Clupea harengus). Mol Ecol. 2012;21:3686–703.
Lal MM, Southgate PC, Jerry DR, Bosserelle C, Zenger KR. Swept away: ocean currents and seascape features influence genetic structure across the 18,000 km indo-Pacific distribution of a marine invertebrate, the black-lip pearl oyster Pinctada margaritifera. BMC Genomics. 2017;18(1):66.
Villacorta-Rath C, Ilyushkina I, Strugnell JM, Green BS, Murphy NP, Doyle SR, et al. Outlier SNPs enable food traceability of the southern rock lobster. Jasus edwardsii Mar Biol. 2016;163(11):223.
Hellberg ME, Burton RS, Neigel JE, Palumbi SR. Genetic assessment of connectivity among marine populations. Bull Mar Sci. 2002;70(1):273–90.
Shanks AL. Pelagic larval duration and dispersal distance revisited. Biol Bull. 2009;216(3):373–85.
Ovenden JR. Crinkles in connectivity: combining genetics and other types of biological data to estimate movement and interbreeding between populations. Mar Freshw Res. 2013;64(3):201–7.
Pérez-Enríquez R, Ávila S, Ibarra AM. Population genetics of the oyster Crassostrea corteziensis in the Gulf of California. Cienc Mar. 2008;34(4):479–90.
Hill AE. A mechanism for horizontal zooplankton transport by vertical migration in tidal currents. Mar Biol. 1991;111:485–92.
Baker P, Mann R. Late stage bivalve larvae in a well-mixed estuary are not inert particles. Estuaries. 2003;26(4):837–45.
Pogson GH. Studying the genetic basis of speciation in high gene flow marine invertebrates. Curr Zool. 2016;62(6):643–53.
Varney RL, Galindo-Sanchez CE, Cruz P, Gaffney PM. Population genetics of the eastern oyster Crassostrea virginica (Gmelin, 1791) in the Gulf of Mexico. J Shellfish Res. 2009;28(4):855–64.
Zainal Abidin DH, Mustaffa S, Rahim MA, Nair DM, Naim DM, Mohd Nor SA. Population genetics of the black scar oyster, Crassostrea iredalei: repercussion of anthropogenic interference. Mitochondrial DNA A. 2016;27(1):647–58.
Launey S, Ledu C, Boudry P, Bonhomme F, Naciri-Graven Y. Geographic structure in the European flat oyster (Ostrea edulis L.) as revealed by microsatellite polymorphism. J Hered. 2002;93:331–8.
Thomson JM. The genera of oysters and the Australian species. Division of Fisheries, CSIRO: Cronulla; 1953.
White MJ. Aspects of growth and reproduction of Saccostrea echinata the indigenous blacklip oyster cultured at Magnetic Island Queensland: James Cook University; 1991.
Southgate PC, Lee PS. Hatchery rearing of the tropical blacklip oyster Saccostrea echinata (Quoy and Gaimard). Aquaculture. 1998;169:275–81.
Nowland SJ, O'Connor WA, Southgate PC. Embryonic, larval, and early postlarval development of the tropical black-lip rock oyster Saccostrea echinata. J Shellfish Res. 2018;37:73–7.
Rose CG, Paynter KT, Hare MP. Isolation by distance in the eastern oyster, Crassostrea virginica, in Chesapeake Bay. J Hered. 2006;97(2):158–70.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95.
Sekino M, Yamashita H. Mitochondrial and nuclear DNA analyses of Saccostrea oysters in Japan highlight the confused taxonomy of the genus. J Molluscan Stud. 2016;82(4):492–506.
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR. NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (ne) from genetic data. Mol Ecol Resour. 2014;14:209–14.
Yapici N, Kim Y-J, Ribeiro C, Dickson BJ. A receptor that mediates the post-mating switch in Drosophila reproductive behaviour. Nature. 2008;451:33–8.
Oh Y, Yoon SE, Zhang Q, Chae HS, Daubnerova I, Shafer OT, et al. A homeostatic sleep-stabilizing pathway in Drosophila composed of the sex peptide receptor and its ligand, the myoinhibitory peptide. PLoS Biol. 2014;12(10):e1001974.
McDougall C. Comparative De novo transcriptome analysis of the Australian black-lip and Sydney rock oysters reveals expansion of repetitive elements in Saccostrea genomes. PLoS One. 2018;13(10):1–15.
Utting SD, Spencer BE. Laboratory leaflet 68: the hatchery culture of bivalve mollusc larvae and juveniles. Lowestoft: Ministry of Agriculture, Fisheries and Food Directorate of Fisheries Research; 1991.
Lucas JS. Bivalve Molluscs. In: Lucas JS, Southgate PC, editors. Aquaculture: Farming Aquatic Animals and Plants. Second Edition ed. Sussex: Blackwell Publishing Ltd; 2012. p. 541–566.
Guo X, Hedgecock D, Hershberger WK, Cooper K, Allen SK. Genetic determinants of protandric sex in the Pacific oyster, Crassostrea gigas Thunberg. Evolution. 1998;52(2):394–402.
Guo X, Li C, Wang H, Xu Z. Diversity and evolution of living oysters. J Shellfish Res. 2018;37(4):755–71.
Guo X, He Y, Zhang L, Lelong C, Jouaux A. Immune and stress responses in oysters with insights on adaptation. Fish Shellfish Immunol. 2015;46(1):107–19.
Kawamura K, Miyake T, Obata M, Aoki H, Komaru A. Population demography and genetic characteristics of the Pacific oyster Crassostrea gigas in Japan. Biochem Syst Ecol. 2017;70:211–21.
Thongda W, Zhao H, Zhang D, Jescovitch LN, Liu M, Guo X, et al. Development of SNP panels as a new tool to assess the genetic diversity, population structure, and parentage analysis of the eastern oyster (Crassostrea virginica). Mar Biotechnol (NY). 2018;20(3):385–95.
Arruda CCB, Beasley CR, Vallinoto M, Marques-Silva NS, Tagliaro CH. Significant genetic differentiation among populations of Anomalocardia brasiliana (Gmelin, 1791): a bivalve with planktonic larval dispersion. Genet Mol Biol. 2009;32(2):423–30.
Bird CE, Holland BS, Bowen BW, Toonen RJ. Contrasting phylogeography in three endemic Hawaiian limpets (Cellana spp.) with similar life histories. Mol Ecol. 2007;16(15):3173–86.
Goodall-Copestake WP, Tarling GA, Murphy EJ. On the comparison of population-level estimates of haplotype and nucleotide diversity: a case study using the gene cox1 in animals. Heredity. 2012;109(1):50–6.
Eckert CG, Samis KE, Lougheed SC. Genetic variation across species' geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol. 2008;17(5):1170–788.
Sexton JP, McIntyre PJ, Angert AL, Ric KJ. Evolution and ecology of species range limits. Annu Rev Ecol Evol Syst. 2009;40:415–36.
Ray N, Currat M, Excoffier L. Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol. 2003;20(1):76–86.
Varney RL, Sackett RE, Wilbur AE. Analysis of spatiotemporal genetic variability in eastern oyster Crassostrea virginica (Gmelin, 1791) Mtdna 16S sequences among North Carolina populations. J Shellfish Res. 2016;35(2):329–42.
Teske PR, Golla TR, Sandoval-Castillo J, Emami-Khoyi A, van der Lingen CD, von der Heyden S, et al. Mitochondrial DNA is unsuitable to test for isolation by distance. Sci Rep. 2018;8(1):8448.
Bowen BW, Shanker K, Yasuda N, Malay MD, Heyden S, Paulay G, et al. Phylogeography unplugged: comparative surveys in the genomic era. Bull Mar Sci. 2014;90(7):13–46.
Nielsen EE, Hemmer-Hansen J, Poulsen NA, Loeschcke V, Moen T, Johansen T, et al. Genomic signatures of local directional selection in a high gene flow marine organism; the Atlantic cod (Gadus morhua). BMC Evol Biol. 2009;9:276.
Helyar SJ, Hemmer-Hansen J, Bekkevold D, Taylor MI, Ogden R, Limborg MT, et al. Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol Ecol Resour. 2011;11:123–36.
Pujolar JM, Jacobsen MW, Als TD, Frydenberg J, Munch K, Jonsson B, et al. Genome-wide single-generation signatures of local selection in the panmictic European eel. Mol Ecol. 2014;23(10):2514–28.
Schiller A. Ocean circulation on the north Australian shelf. Cont Shelf Res. 2011;31(10):1087–95.
Geoscience Australia. Commonwealth of Australia; 2018 [Available from: http://www.ga.gov.au/scientific-topics/energy/province-sedimentary-basin-geology/petroleum/offshore-northwest-australia/arafura-basin#heading-1. Accessed 18 Sept 2018.
Monash University. Sahul Time: Monash University; 2007 [Available from: http://sahultime.monash.edu.au/.
Gardner MG, Li X, Fitch AJ. Population genetic structure of sea cucumbers (bêche-de-mer) in northern Australia. The Seafood CRC; 2012.
Grosjean E, Logan GA, Rollet N, Ryan GJ, Glenn K. Geochemistry of shallow tropical marine sediments from the Arafura Sea. Australia Org Geochem. 2007;38:1953–71.
Kim YJ, Bartalska K, Audsley N, Yamanaka N, Yapici N, Lee JY, et al. MIPs are ancestral ligands for the sex peptide receptor. Proc Natl Acad Sci U S A. 2010;107(14):6520–5.
Poels J, Van Loy T, Vandersmissen HP, Van Hiel B, Van Soest S, Nachman RJ, et al. Myoinhibiting peptides are the ancestral ligands of the promiscuous Drosophila sex peptide receptor. Cell Mol Life Sci. 2010;67(20):3511–22.
DoF. Abalone aquaculture in Western Australia: Principles and considerations relating to management of abalone aquaculture in Western Australia. 2016. Contract No.: 132.
Galvão MSN, Pereira OM, Hilsdorf AWS. Molecular identification and distribution of mangrove oysters (Crassostrea) in an estuarine ecosystem in Southeast Brazil: implications for aquaculture and fisheries management. Aquac Res. 2013;44(10):1589–601.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol. 2017;34:3299–302.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10:564–7.
Dray S, Dufour A. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22(4):1–20.
Bandelt H, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48.
Leigh JW, Bryant D. PopART: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6(9):1110–6.
Suzana M, Mohd Lutfi A, Abdul Hadi A, Devakie MN, Siti Azizah MN. Genetic variation in Malaysian oysters: taxonomic ambiguities and evidence of biological invasion. Biol Invasions. 2011;13(8):1893–900.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Rambaut A. FigTree v1.4.4. Edinburgh: University of Edinburgh; 2006.
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7(5):e37135.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22(11):3124–40.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Lischer HEL, Excoffier L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 2012;28:298–9.
Foll M, Gaggiotti OE. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–93.
Meirmans PG, Van Tienderen PH. GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes. 2004;4:792–4.
R Core Team. R: A language and environment for statistical computing. Vienna: The R Foundation; 2017.
Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94.
Berry O, Tocher MD, Sarre D. Can assignment tests measure dispersal? Mol Ecol. 2004;13:551–61.
Paetkau D, Slade R, Burden M, Estoup A. Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol. 2004;13(1):55–65.
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M. New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics. 1999;153:1989–2000.
The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2014;43:204–12.
Lam K, Morton B. Morphological and mitochondrial-DNA analysis of the indo-West Pacific rock oysters (Ostreidae: Saccostrea species). J Molluscan Stud. 2006;72(3):235–45.
Acknowledgements
We wish to thank the Darwin Aquaculture Centre, Bowen Fresh Oysters, the Government of Western Australia, the Pacific Community, and the following Aboriginal organisations; Yagbani Aboriginal Corporation, Tiwi Land Council, Tiwi Marine Rangers, Munupi Family Trust, Crocodile Island Rangers, Dhimurru Rangers, Yirrkala Rangers, and Anindilyakwa Rangers for their support and assistance with this project. We thank Professor Wayne O’Connor, Dr. Shane Penny and the reviewers and editors who made valuable contributions to this manuscript.
Funding
This study was conducted within the Australian Northern Territory Government “Tropical Rock Oyster Aboriginal Economic Development Program” that was administered by the Darwin Aquaculture Centre, Department of Primary Industry and Resources of the Northern Territory Government. The Program scientist (Samantha Nowland) is the first author of this paper and was involved in all aspects of the research; design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
SJN carried out all tissue collections, participated in the investigation design and conceptualisation, performed data analyses, drafted the manuscript and provided project funding. CNSS carried out the DNA extraction, purification and optimised samples prior to library preparation, assisted with data analyses and interpretation, and edited the manuscript. PCS developed the broad project concept, participated in the investigation design, provided supervisory support, advice on tropical oyster biology and ecology, and edited the manuscript. JMS participated in design and conceptualisation of the project, provided statistical advice and technical input on investigation design, assisted with data analyses and interpretation, provided supervisory support and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The work described herein has been carried out (where appropriate) in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for animal experiments. All oysters were handled in accordance with the Northern Territory Governments animal ethics requirements and guidelines.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
This file contains a table of Kimura 2-parameter genetic distances in mitochondrial COI sequence data. (DOCX 13 kb)
Additional file 2:
Phylogenetic analysis of Saccostrea COI sequences. Bootstrap values are given on branches, and the scale bar indicates the number of substitutions per site. The clades containing Striostrea, Magallana and Crassostrea COI sequences are used as outgroups. Lineages have been designated (where possible) following Lam and Morton [86] and Sekino and Yamashita [32]. (PDF 386 kb)
Additional file 3:
This file contain mitochondrial COI sequence data accession numbers for 273 individuals of S. echinata. (XLSX 13 kb)
Additional file 4:
This VCF file contains genotypic data of 272 individuals of S. echinata at 27,887 genome-wide SNPs. (ZIP 12536 kb)
Additional file 5:
This file includes a list of identifying numbers and sequences for the 31 candidate adaptive SNPs. (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nowland, S.J., Silva, C.N.S., Southgate, P.C. et al. Mitochondrial and nuclear genetic analyses of the tropical black-lip rock oyster (Saccostrea echinata) reveals population subdivision and informs sustainable aquaculture development. BMC Genomics 20, 711 (2019). https://doi.org/10.1186/s12864-019-6052-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6052-z