
Abstract
Background
Aquaporins (AQPs) facilitate transport of water and small solutes across cell membranes and play an important role in different physiological processes in plants. Despite their importance, limited data is available about AQP distribution and function in the economically important oilseed crop peanut, Arachis hypogea (AABB). The present study reports the identification and structural and expression analysis of the AQPs found in the diploid progenitor genomes of A. hypogea i.e. Arachis duranensis (AA) and Arachis ipaensis (BB).
Results
Genome-wide analysis revealed the presence of 32 and 36 AQPs in A. duranensis and A. ipaensis, respectively. Phylogenetic analysis showed similar numbers of AQPs clustered in five distinct subfamilies including the plasma membrane intrinsic proteins (PIPs), the tonoplast intrinsic proteins (TIPs), the nodulin 26-like intrinsic proteins (NIPs), the small basic intrinsic proteins (SIPs), and the uncharacterized intrinsic proteins (XIPs). A notable exception was the XIP subfamily where XIP1 group was observed only in A. ipaensis genome. Protein structure evaluation showed a hydrophilic aromatic/arginine (ar/R) selectivity filter (SF) in PIPs whereas other subfamilies mostly contained a hydrophobic ar/R SF. Both genomes contained one NIP2 with a GSGR SF indicating a conserved ability within the genus to uptake silicon. Analysis of RNA-seq data from A. hypogea revealed a similar expression pattern for the different AQP paralogs of AA and BB genomes. The TIP3s showed seed-specific expression while the NIP1s’ expression was confined to roots and root nodules.
Conclusions
The identification and the phylogenetic analysis of AQPs in both Arachis species revealed the presence of all five sub-families of AQPs. Within the NIP subfamily, the presence of a NIP2 in both genomes supports a conserved ability to absorb Si within plants of the genus. The global expression profile of AQPs in A. hypogea revealed a similar pattern of AQP expression regardless of the subfamilies or the genomes. The tissue-specific expression of AQPs suggests an important role in the development and function of the respective organs. The AQPs identified in the present study will serve as a resource for further characterization and possible exploitation of AQPs to understand their physiological role in A. hypogea.
Similar content being viewed by others


Background
Aquaporins (AQPs) are small (21–34 kD) integral membrane proteins, which form channels facilitating movement of water and other small solutes across the cell membrane. Aquaporins are conspicuously present across all kingdoms of life including plants where they co-ordinate water transport from the soil to different plant parts [1,2,3,4]. Based on sequence similarity and subcelluar localization, five subfamilies of AQPs have been identified in seed plants: the plasma membrane intrinsic proteins (PIPs), the tonoplast intrinsic proteins (TIPs), the nodulin26-like intrinsic proteins (NIPs), the small basic intrinsic proteins (SIPs) and the uncategorized intrinsic proteins (XIPs) [5,6,7,8]. Variation in the number of AQP subfamilies specific to different plant species has been reported. Among the five subfamilies, XIPs are absent entirely from monocots and dicots like Brassicaceae [9,10,11]. In primitive land plants, two additional unique classes of AQPs, GlpF-like intrinsic protein (GIPs) and hybrid intrinsic proteins (HIPs) have been described and are presumed to have been lost in the course of evolution [12]. Among AQPs, TIPs and PIPs are specifically located in vacuolar and plasma membranes, respectively. Being the most abundant in plants, TIPs and PIPs play a central role in mediating water transport across the plant system. The SIPs were the first to be unraveled via genome sequence analysis and are generally localized in the endoplasmic reticulum (ER). NIPs are homologous to GmNod26, an abundantly expressed transcript in the peribacteroid membrane of nitrogen-fixing nodules of soybean roots [13] and are mostly found in the plasma membrane.
The general AQP structure resembles an hourglass formed by six transmembrane (TM) α helices (H1 to H6) joined by five inter-helical loops (A to E). At the center of the pore formed by the six TM domains, two different constricts are formed: one that harbors conserved NPA (Asn-Pro-Ala) motifs, and another one known as aromatic/arginine (ar/R) selectivity filter (SF) formed with four amino acids in the channel. Among the four amino acids, one is located in each of helix 2 (H2) and helix 5 (H5), and two residues are located in loop E (LE1 and LE2). These two constrictions predominantly determine solute specificity and permeability within a given AQP [9, 14, 15].
The availability of whole genome sequences in cultivated crop plants has accelerated the genome-wide identification and analysis of the AQP-encoding genes [16]. The genome-wide characterization of AQPs has revealed important properties such as their distribution, evolution and conserved structural features involved in solute transport [16]. In this context, identification and characterization of AQP genes is the first step to decipher their presence and role in regulating transport of water and other physiologically important molecules. Translating this information to crop plants carries important implications with regards to breeding or engineering plants with improved water and nutrient uptake.
Plant AQPs exhibit abundant diversity in comparison with AQPs from bacteria and animals. This assists plants to overcome their disadvantage of immobility as they encounter varied environmental and climatic conditions. While initially AQPs were largely known as water channel proteins, they are now recognized to transport a plethora of small solutes like urea, H2O2, silicon, boron, ammonia and CO2 [17]. The regulation of AQP genes in response to biotic and abiotic stresses has been reported in several crop plants [18,19,20]. Aquaporins also serve as key regulators modulating plant growth and development during various physiological and environmental states.
Arachis hypogea (L.), popularly known as peanut, is by far the most economically important species of the Arachis genus. It is an allotetraploid (2n = 4x = 40), thought to be derived from a single recent hybridization event between two wild ancestors, Arachis duranensis (AA) and Arachis ipaensis (BB) [21]. The crop is valued for the kernel, an important source of protein (28%), edible oil (42%), and numerous nutrients and minerals [22]. The production of the A. hypogea can be altered by different biotic and abiotic stresses causing significant yield losses annually. In recent years, weather fluctuations have caused severe water-deficit conditions threatening the sustainable production of A. hypogea. Drought causes tissue dehydration due to an imbalance between plant water uptake and transpiration [23]. These imbalances can be alleviated by AQPs, which play an important role in maintaining water balance and homeostasis under different environmental and stress conditions [24]. However, very little is known about the AQP distribution and function in A. hypogea (AABB), and how they could help efforts to develop more drought tolerant cultivars .
Recently the two progenitor genomes, A. duranensis and A. ipensis were sequenced to facilitate the study of the complete genome of cultivated A. hypogea [21]. In the present study, we took advantage of these available sequences to identify all AQPs in the diploid progenitor genomes of A. hypogea. Subsequently, we were able to characterize them according to their phylogenetic distribution, gene structure, conserved motifs and ar/R SF. Finally, we analyzed AQP expression in different tissues using available transcriptomic data from A. hypogea. This study brings novel and relevant information with regards to the many and specific functions AQPs play in A. hypogea and offers avenues to exploit this information to improve stress resistance in A. hypogea.
Results
Genome-wide identification and distribution of AQPs in A. duranensis and A. ipaensis
The homology based search performed in the A. duranensis and A. ipaensis genomes revealed the presence of 32 and 36 AQPs, respectively. Subsequent identification of conserved domains also confirmed all the predicted AQPs (Additional file 1). Interestingly, based on the recent release of A. hypogea genome, we observed 73 AQPs distributed among different subfamilies (Additional file 2). HiddenMarkov model-based prediction of transmembrane helices showed the presence of six signature transmembrane domains in 23 out of 32 AQPs in A. duranensis and 23 out of 36 in A. ipaensis (Additional file 3). Tertiary protein structure analysis of the AQPs confirmed the typical hourglass-like structure formed with six TM domains for all proteins analysed except AipNIP1–3 (Additional file 4). The A. duranensis and A. ipaensis AQPs were found to be distributed among nine out of 10 chromosomes. In A. duranensis, the highest number (six) of AQPs were found on chromosome 3, 9 and 10 (Table 1). Similarly, in A. ipaensis the highest number (10) of AQPs was found on chromosome 3, while five AQPs each were found on chromosome 9 and 10 (Table 2).
Phylogenetic and gene structure analysis of AQPs in A. duranensis and A. ipaensis
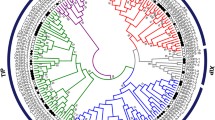
Phylogenetic analysis of AQP candidates from A. duranensis and A. ipaensis along with known AQPs from Arabidopsis thaliana and Glycine max formed five distinct clusters representing different classes of AQPs (Fig. 1). The AQP candidates were classified according to their respective cluster. In A. duranensis, AQPs were grouped into nine PIPs, 11 TIPs, eight NIPs, three SIPs and one XIP. For their part, AQP candidates from A. ipaensis were grouped into nine PIPs, 10 TIPs, 10 NIPs, three SIPs and four XIPs. The AduPIPs and AipPIPs formed two major sub-groups of PIP1s and PIP2s comprising five and four members, respectively. Likewise, the TIP family classified into five subclusters containing a different number of TIPs in each subcluster. NIPs formed three (NIP1, NIP2 and NIP3) and four (NIP1, NIP2 NIP3 and NIP4) groups in A. duranensis and A. ipaensis respectively. The SIPs from both species formed two groups, SIP1 and SIP2, containing two and one members, respectively. Among XIPs, no XIP1 was found and only a single member of XIP2 (AduXIP2–1) was observed in A. duranensis. In A. ipaensis, XIP1s had three members (AipXIP1–1, AipXIP1–2and AipXIP1–3) and XIP2s (AipXIP2–1) had one member.

Phylogenetic tree of Arachis duranensis and Arachis ipaensis aquaporins along with Arabidopsis thaliana and Glycine max. The analysis grouped aquaporins into five different clusters. The genes from A. duranensis, A. ipaensis, A. thaliana and G.max are preceded by the prefixes Adu, Aip, At, and Gm, respectively. The number next the branches represents bootstrap values ≥50% based on 1000 resamplings
Aquaporins from both species displayed less variation in CDS length (A. durensis: 564 bp to 939 bp; A. ipaensis: 531 bp to 1053 bp) than in gene length (A. durensis: 650 bp to 10,326 bp; A. ipaensis: 750 bp to 8695 bp). Gene structure analysis revealed considerable variation in both number and length of introns and exons that resulted in gene length variation (Fig. 2). In A. duranensis, the number of introns varied from one (AduTIP1–1, AduTIP1–2, AduTIP1–3, AduTIP2–1) to 4 (AduNIP1–1, AduNIP1–2, AduNIP1–3, AduNIP1–5, AduNIP2–1, AduNIP3–1), while in A. ipaensis, this number varied from one (AipTIP1–1, AipXIP1–1, AipXIP1–2, AipNIP3–3) to six (AipNIP2–1).

Exon–intron organization of aquaporin genes identified in genomes of (a) Arachis duranensis and (b) Arachis ipaensis. Graphical output of the gene model was obtained using Gene Structure Display Server (http://gsds.cbi.pku.edu.cn/). Exons are shown as geen boxes and introns are shown as black lines. The scale shown at the bottom reperesents gene length in base pairs
Characterization of NPA motif, transmembrane domains and sub-cellular localization of A. duranensis and A. ipaensis AQPs
Candidate Arachis AQPs were found to have differences in NPA motifs and residues at ar/R SF (Table 3 and Table 4). In both species, all PIPs and TIPs displayed two conserved NPA motifs. Among NIPs, NIP1s and NIP2s showed two conserved NPA domains, while NIP3s showed variation. Among NIP3s the variation included a substitution of alanine by serine or valine and substitution of asparagine by lysine. Additionally, all the members of the XIP and SIP subfamilies had varying NPA domains. All PIP subfamily members from both studied species displayed conservation at the ar/R SF residues (Table 3 and Table 4) with phenylalanine at H2, histidine at H5, threonine at LE1 and arginine at LE2 (Table 3 and Table 4). Most of the TIP subfamily members showed group specific conservation of ar/R SF. For instance, all TIP1s contained Histidine-Isoleucine-Alanine-Valine, while all TIP2s comprised of Histidine-Isoleucine-Glycine-Arginine in both the species. Similarly, NIPs also displayed subgroup specific conservation of ar/R SF except NIP3s, which displayed variation in the SF.
To characterize spatial expression of AQPs from both species, their subcellular localizations were predicted in silico (Additional file 5). In A. duranensis and A. ipeanensis, the majority of the PIP homologs were predicted to be localized in the plasma membrane but AduPIP1–4 that was predicted to be localized in the mitochondria. Expectedly, most TIP sub-family members were predicted to be in the vacuole. However, a few family members were predicted to be localized in the cytoplasm (AduTIP1–1, AipTIP3–1), the chloroplast (AduTIP1–2, AduTIP5–1, AipTIP1–3, AipTIP5–1), Mitochondria (AduTIP3–1) and the plasma membrane (AipTIP2–2). Most NIPs from both species were expected to be found in the plasma membrane. The SIP family members had candidates in different sites including the plasma membrane (AduSIP1–1, AipSIP1–1, AipSIP1–2), the chloroplast (AduSIP1–2) and the cytoplasm (AduSIP2–1, AipSIP2–1). The members of XIPs were found to be likely localized in the cytoplasm, chloroplast, or nucleus.
Aquaporin expression profiling across different tissues in A. hypogea
Analysis of RNA-seq data from A. hypogea showed expression of 27 out of 32, and 32 out of 36 identified AQPs from A. duranensis and A. ipeanensis, respectively. Similar patterns of expression for members of different AQP subfamilies from both AA and BB genomes were observed. Analysis of expression of AQPs at different developmental stages revealed a higher expression of PIPs followed by TIPs, SIPs, NIPs and XIPs (Fig. 3). In general, PIPs showed higher expressions across all tissues analyzed. Among TIPs, TIP2s (AduTIP2–2 and AipTIP2–3) showed higher expression in the roots, pistil and leaves. Higher expression of TIP3s (AduTIP3–1 and AipTIP3–1) was observed in all the five different developmental stages of seeds. Similarly, few NIPs (AduNIP1–2, AipNIP1–3) showed high to moderate expression in four out of five different developmental stages of seeds. The AduNIP1–4 and AipNIP1–5 showed strong expression in root nodules. Among XIPs, no expression of the unique XIP member specific to the AA genome (AduXIP2–1) was observed. However, the BB genome specific XIP members, AipXIP2–1 accumulated at higher levels in nodules of A. hypogea.

Analysis of the expression of Arachis hypogea aquaporins in different tissues using RNA-seq data (PRJNA291488, BioProject). Normalized expression of aquaporins in terms of reads per kilobase of transcript per million mapped reads (RPKM) showing higher levels of PIP and TIP expression compared to NIP and XIP expression across the different tissues analyzed
Discussion
In this study, we exploited the availability of the whole genome sequence of A. duranensis and A. ipeansis, the progenitors of cultivated A. hypogea, [21] to provide an exhaustive identification and characterization of A. hypogea AQPs as a way to facilitate a better understanding of their roles in the development of the plant. Although the full genome of A. hypogea has recently been made available, genome-wide study of AQPs in true diploid progenitor species provides some advantages over its analysis in a highly complex and polyploid genome like peanut. Indeed, it is more informative about the relative importance and function of each aquaporin in its respective diploid progenitor, and about the impact of genome polyploidization on AQP gene structure, function and dosage-dependence on gene expression pattern. The advent of next generation sequencing platforms has enabled the decoding of AQPs in many plant species [25,26,27] and has highlighted their many functions in metabolism regulation, namely in the case of biotic and abiotic stresses [28], information that can have many positive implications in developing new varieties better adapted to stress conditions.
The number of AQPs identified in A. duranensis and A. ipaensis, 32 and 36, was found to be fairly proportional to their respective genome size of 1.25 and 1.56 Gb [21]. By comparison, many dicots such as Arabidopsis (35) [7], Phaseolus (41) [29], and pigeon pea (40) [30] bear similar numbers. On the other hand, some plant species such as canola which evolved with polyploidization contains as many as 120 AQPs [28]. However, notwithstanding this lower number of AQP candidates, the phylograms of both species showed homologs representing all five subfamilies (PIPs, TIPs, NIPs, SIPs and XIPs) as observed in most higher plant species. The presence of XIPs is particularly interesting since all monocots and dicots belonging to the Brassicaceae family are characterized by a complete absence [6, 7, 27]. The analysis of A. hypogea genome revealed the presence of 73 AQPs representing homologs of most of the AQPs identified from its progenitor genomes, A. duranensis (32) and A. ipaensis (36). The difference in the number of aquaporin genes in A. hypogea can be attributed to gene duplication and loss specific to different subfamilies of AQPs over the course of evolution. Similar observations of gene expansion have been reported in LEA and SWEET genes in Brassica napus (AACC) compared to its progenitors, Brassica rapa (AA) and Brassica oleracea (CC) [31, 32]. The exon-intron structure observed in the two Arachis species was found conserved and does correlate well with their phylogenetic distribution. Since the exon-intron structure in AQP subfamilies was similar in both species, this indicates that a diversification of AQPs preceded the evolution of the genus Arachis. The similar gene structure also points to a conserved function of AQPs within the genus. The intron number was reported to be correlated to gene expression, duplication, and diversification in plants [33]. For instance, the high intron number variation in NIPs is correlated with their vulnerability to evolution.
The two conserved NPA motifs along with the four amino acids that form the ar/R SF largely determine solute specificity and transport of the substrate across AQPs [9, 14, 15]. Based on our analyses, the respective members of each AQP subfamily in both Arachis species showed conserved NPA motifs and similar ar/R SF. Indeed, all PIPs were found to harbor the characteristic double NPA motif and a hydrophilic ar/R SF (F/H/T/R) as observed in AqpZ [34], confirming their affinity to transport water. The same filter was found conserved for PIP members from other plant species such as Zea mays [5], Solanum lycopersicum [35], A. thaliana [7], G. max [30], and Phaseolus vulgaris [29]. PIP members are known to regulate water transport in several plant species and play an instrumental role in maintaining root and leaf hydraulics [29]. PIPs have also been shown to regulate photosynthesis in A. thaliana, Hordeum vulgare and Nicotiana tabaccum by facilitating CO2 diffusion in mesophyll tissues [29]. Therefore, the conserved features of PIPs suggest similar functions in Arachis, a conclusion reinforced by RNA-seq analyses that showed a higher expression of PIPs across different tissues analyzed.
Among TIPs, NPA motifs were conserved and, TIP1s and TIP3s showed more hydrophobic residues than TIP2s, TIP4s and TIP5s. Generally, TIPs are located in vacuolar membranes and act as transporters of water and small solutes like ammonia, hydrogen peroxide, boron and urea [36,37,38,39]. The residues found in the ar/R SF in TIP subfamilies were similar to TIPs from other plant species pointing to a similar conserved role in Arachis species. A high accumulation of TIP3s was observed in seeds from different developing stages of A. hypogea, a phenomenon observed in A. thaliana [40, 41], H. vulgare [42] and G. max [30] and reported as a role in seed desiccation processes. The TIP3s are involved in maturation of the vacuolar apparatus and allow optimal water uptake during embryo development and seed germination [43]. Recently, BvCOLD1, a boron transport TIP was found to be involved in cold tolerance in sugar beet [39]. Further studies on TIP regulation could help better understand why cold stress represents a major limitation for peanut cultivation. Interestingly, in a recent study, Devi et al. [44] suggested that putative TIPs and PIPs in A. hypogea played a role in regulating drought tolerance.
Among the NIPs, NIP1s showed a selectivity filter with more hydrophobicity (WVAR) compared to NIP2s and NIP3s. In the present study, a single NIP2 gene containing a GSGR selectivity filter was observed in both Arachis species. NIP2s with a GSGR selectivity filter play a unique role in plants by allowing influx of silicon (Si) [9, 30]. In turn, Si accumulation has been shown to protect plants against a wide variety of biotic and abiotic stresses [45]. Interestingly, the presence of functional NIP2s for Si permeability vary greatly among plant species and our results bring the first evidence that A. hypogea has the appropriate channel to benefit from Si fertilization. In general, NIPs will display lower expression than PIPs or TIPs, and our results confirmed this trend whereAduNIP1–4 and AipNIP1–5 were found specifically expressed in roots and root nodules. A similar specificity of NIP expression was observed in G. max [46] and Medicago truncatula [47]. Additionally, in M. truncatula, NIPs were found to be expressed from the early to late stage of nodule development, which indicates their importance in nodule organogenesis [47].
In the XIP subfamily, a single XIP2 member was found in A. duranensis, while members of both XIP1s and XIP2s were observed in A. ipaensis. This suggests that A. duranensis lost XIP1s during the course of evolution, an observation reported in many other species including all monocots, which raises the question of their importance or role in plants. When they are present, the hydrophobic nature of their selectivity filter is believed to facilitate the transport of hydrophobic and bulky molecules such as urea, glycerol, and boric acid in plants [48]. Interestingly, the expression data showed that the only A. ipaensis XIP member, AipXIP2–1, accumulated at higher levels in the nodules of A. hypogea supporting its involvement in nodule development. Several studies have established the role of AQPs in key developmental processes [49, 50]. For instance, it has been reported that the increased abundance of TIPs facilitates the development of new lateral root primordia in A. thaliana [51]. Nevertheless, XIPs deserve further studies since their exact role and the consequences of their loss in some species, remain poorly understood.
Conclusions
Genome-wide analysis and characterization of the AQP gene family were performed in A. duranensis (AA) and A. ipaensis (BB) the probable progenitor genomes of A. hypogea (AABB). The identification and the phylogenetic analysis of AQPs in both species revealed the presence of all five sub-families of AQPs. Within the XIP subfamily, the loss of XIP1s from the AA genome was observed, while the presence of a NIP2 in both genomes support a conserved ability to absorb Si within plants of the genus. The global expression profile of AQPs in A. hypogea through RNA-seq data analysis revealed a similar pattern of expression of AQPs regardless of the subfamilies or the genomes. A higher expression of TIP3s was observed in different stages of seed development in A. hypogea supporting a critical physiological role of TIP3s in seed development. The high accumulation of the BB genome specific-AipXIP2–1 in nodules of A. hypogea suggests a novel role in nodule development for the elusive XIPs. The AQPs identified in the present study will serve as a resource for further characterization and possible exploitation of AQPs to understand their physiological role in A. hypogea.
Methods
Genome-wide identification and distribution of AQPs in A. duranensis and A. ipaensis
The genome sequences of A. duranensis A. ipaensis and A. hypogea were retrieved from the PeanutBase (https://peanutbase.org/) [52]. Predicted protein sequences were used to create a local database using BioEdit ver. 7.2.5 [53]. Homologs of the AQPs coding genes were identified by BLASTp search performed against the local database using AQPs from A. thaliana, Oryza sativa and G. max (Additional file 6). An e-value of 10− 5 was kept as an initial cut-off to identify high scoring pairs (HSPs). The blast output was tabulated, and the HSPs having greater than 100-bit score were selected. Finally, redundant hits were removed to select unique sequences for further analysis.
Structural characterization of A. duranensis and A. ipaensis AQPs
The genomic and cDNA sequences of AQPs identified in A. duranensis and A.ipaensis were retrieved from PeanutBase. Structural annotations of the gene models (in gff3 format) were also retrieved from PeanutBase. The gene structure of AQPs was analyzed using GSDS ver. 2.0 [54].
Identification of functional motif and transmembrane domains and estimation of isoelectric point (pI) for AQPs
The NPA motifs were identified in predicted protein sequences using conserved domain database (CDD, www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) [55]. Missing NPA motifs in few AQP sequences were confirmed with a manual examination. Transmembrane domains in the genes were identified using TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) [56] and SOSUI online software tools [57]. The transmembrane domains were analyzed manually to confirm alterations or complete loss. The isoelectric point (pI) of AQP protein sequences were calculated using the online tool Sequence Manipulation Suite version 2 (http://www.bioinformatics.org/sms2/protein_iep.html) [58].
Phylogenetic analysis of AQPs in A. duranensis and A. ipaensis
The predicted AQP protein sequences were aligned using CLUSTALW alignment tool in MEGA6 [59]. The phylogenetic tree was constructed by the maximum likelihood method, and the stability of the branch node was analysed by performing 1000 bootstraps. The AQP subfamilies, PIP, SIP, TIP, NIP, and XIP, were classified according to the nomenclature used for A. thaliana, and G. max [6, 9].
Tertiary protein structure prediction
The tertiary (3D) protein structure of A. duranensis and A. ipaensis AQPs were generated using the Phyre2 protein-modeling server (http://www.sbg.bio.ic.ac.uk/~phyre2) [60] with extensive mode. Identification of transmembrane pore, pore lining residues, pore morphology and constricts in the 3D protein structures were performed using PoreWalker server (http://www.ebi.ac.uk/thornton-srv/software/PoreWalker/) [61].
Expression profiling of A. hypogea AQPs
RNA-seq data derived from 22 different tissues of cultivated peanut available in PeanutBase (Genbank BioProject PRJNA291488) were used for expression analysis. The transcriptome assembly and expression value estimation were done as described in Clevenger et al. [62]. Briefly, de novo assembly was carried out by a genome-guided approach using assembly pipeline from Trinity [63]. Total reads were mapped to the transcript assembly from 58 libraries using Bowtie [64], allowing two mismatches within a particular 25 bp seed. Uniquely mapped raw read counts per gene were normalized using the formula of Reads Per Kilobase of transcript per Million mapped reads (RPKM) = Number of Reads / (Gene Length/1000 * Total Number of Reads/1,000,000). The RPKM values for AQPs were extracted and used for heat map preparation. A heat map was constructed using TIGR Multi Experiment Viewer (MeV,http://mev.tm4.org). Hierarchical clustering with average linkage method was performed to cluster the AQPs.
Abbreviations
- AQP:
-
Aquaporin
- ar/R:
-
Aromatic/arginine
- NIP:
-
Nodulin26-like Intrinsic Proteins
- NPA:
-
Asparagine-proline-alanine
- PIP:
-
Plasma membrane Intrinsic Protein
- SF:
-
Selectivity Filter
- SIP:
-
Small basic Intrinsic Proteins
- TIP:
-
Tonoplast Intrinsic Protein
- TM:
-
Transmembrane helix
- XIP:
-
X Intrinsic Proteins
References
Suga N, Takada H, Nomura A, Ohga S, Ishii E, Ihara K, Ohshima K, Hara T. Perforin defects of primary haemophagocytic lymphohistiocytosis in Japan. Br J Haematol. 2002;116:346–9.
Lian H-L, Yu X, Ye Q, Ding X-S, Kitagawa Y, Kwak S-S, Su W-A, Tang Z-C. The role of aquaporin RWC3 in drought avoidance in rice. Plant Cell Physiol. 2004;45:481–9.
Maurel C, Boursiac Y, Luu D-T, Santoni V, Shahzad Z, Verdoucq L. Aquaporins in plants. Physiol Rev. 2015;95:1321–58.
Maurel C, Verdoucq L, Luu DT, Santoni V. Plant aquaporins: membrane channels with multiple integrated functions. Annu Rev Plant Biol. 2008;59:595–624.
Chaumont F, Barrieu F, Wojcik E, Chrispeels MJ, Jung R. Aquaporins constitute a large and highly divergent protein family in maize. Plant Physiol. 2001;125:1206–15.
Quigley F, Rosenberg JM, Shachar-Hill Y, Bohnert HJ. From genome to function: the Arabidopsis aquaporins. Genome Biol. 2002;3:1–17.
Johanson U, Karlsson M, Johansson I, Gustavsson S, Sjövall S, Fraysse L, Weig AR, Kjellbom P. The complete set of genes encoding major intrinsic proteins in Arabidopsis provides a framework for a new nomenclature for major intrinsic proteins in plants. Plant Physiol. 2001;126:1358–69.
Kaldenhoff R, Fischer M. Functional aquaporin diversity in plants. Biochim Biophys Acta. 1758;2006:1134–41.
Deshmukh RK, Vivancos J, Ramakrishnan G, Guérin V, Carpentier G, Sonah H, Labbé C, Isenring P, Belzile FJ, Bélanger RR. A precise spacing between the NPA domains of aquaporins is essential for silicon permeability in plants. Plant J. 2015;83:489–500.
Gupta AB, Sankararamakrishnan R. Genome-wide analysis of major intrinsic proteins in the tree plant Populus trichocarpa: characterization of XIP subfamily of aquaporins from evolutionary perspective. BMC Plant Biol. 2009;9:134.
Shivaraj SM, Deshmukh R, Bhat JA, Sonah H, Bélanger RR. Understanding Aquaporin Transport System in Eelgrass (Zostera marina L.), an Aquatic Plant Species. Front Plant Sci. 2017;8:1334.
Danielson JA, Johanson U. Unexpected complexity of the aquaporin gene family in the moss Physcomitrella patens. BMC Plant Biol. 2008;8:45.
Fortin MG, Morrison NA, Verma DPS. Nodulin-26, a peribacteroid membrane nodulin is expressed independently of the development of the peribacteroid compartment. Nucleic Acids Res. 1987;15:813–24.
Törnroth-Horsefield S, Wang Y, Hedfalk K, Johanson U, Karlsson M, Tajkhorshid E, Neutze R, Kjellbom P. Structural mechanism of plant aquaporin gating. Nature. 2006;439:688–94.
Lee JK, Kozono D, Remis J, Kitagawa Y, Agre P, Stroud RM. Structural basis for conductance by the archaeal aquaporin AqpM at 1.68 Å. PNAS. 2005;102:18932–7.
Deshmukh RK, Sonah H, Bélanger RR. Plant Aquaporins: genome-wide identification, transcriptomics, proteomics, and advanced analytical tools. Front Plant Sci. 2016;7:1896.
Chaumont F, Tyerman SD. Plant Aquaporins: from transport to signaling: Springer; 2017.
Alexandersson E, Fraysse L, Sjövall-Larsen S, Gustavsson S, Fellert M, Karlsson M, Johanson U, Kjellbom P. Whole gene family expression and drought stress regulation of aquaporins. Plant Mol Biol. 2005;59:469–84.
Jang JY, Kim DG, Kim YO, Kim JS, Kang H. An expression analysis of a gene family encoding plasma membrane aquaporins in response to abiotic stresses in Arabidopsis thaliana. Plant Mol Biol. 2004;54:713–25.
Zhou S, Hu W, Deng X, Ma Z, Chen L, Huang C, Wang C, Wang J, He Y, Yang G. Overexpression of the wheat aquaporin gene, TaAQP7, enhances drought tolerance in transgenic tobacco. PLoS One. 2012;7:e52439.
Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EK, Liu X, Gao D, Clevenger J, Dash S. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. 2015;47:438–46.
Özcan MM. Some nutritional characteristics of kernel and oil of peanut (Arachis hypogaea L.). J Oleo Science. 2010;59:1–5.
Sharma KK, Lavanya M: Recent developments in transgenics for abiotic stress in legumes of the semi-arid tropics. JIRCAS Working Report No 23 2002; 23:61–73.
Luu DT, Maurel C. Aquaporins in a challenging environment: molecular gears for adjusting plant water status. Plant Cell Environ. 2005;28:85–96.
Sakurai J, Ishikawa F, Yamaguchi T, Uemura M, Maeshima M. Identification of 33 rice aquaporin genes and analysis of their expression and function. Plant Cell Physiol. 2005;46:1568–77.
Tao P, Zhong X, Li B, Wang W, Yue Z, Lei J, Guo W, Huang X. Genome-wide identification and characterization of aquaporin genes (AQPs) in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol Gen Genomics. 2014;289:1131–45.
Diehn TA, Pommerrenig B, Bernhardt N, Hartmann A, Bienert GP. Genome-wide identification of aquaporin encoding genes in Brassica oleracea and their phylogenetic sequence comparison to Brassica crops and Arabidopsis. Front Plant Sci. 2015;6:166.
Sonah H, Deshmukh RK, Labbé C, Bélanger RR. Analysis of aquaporins in Brassicaceae species reveals high-level of conservation and dynamic role against biotic and abiotic stress in canola. Sci Rep. 2017;7.
Ariani A, Gepts P. Genome-wide identification and characterization of aquaporin gene family in common bean (Phaseolus vulgaris L.). Mol Gen Genomics. 2015;290:1771–85.
Deshmukh RK, Vivancos J, Guerin V, Sonah H, Labbe C, Belzile F, Belanger RR. Identification and functional characterization of silicon transporters in soybean using comparative genomics of major intrinsic proteins in Arabidopsis and rice. Plant Mol Biol. 2013;83:303–15.
Liang Y, Xiong Z, Zheng J, Xu D, Zhu Z, Xiang J, Gan J, Raboanatahiry N, Yin Y, Li M. Genome-wide identification, structural analysis and new insights into late embryogenesis abundant (LEA) gene family formation pattern in Brassica napus. Sci Rep. 2016;6:24265.
Jian H, Lu K, Yang B, Wang T, Zhang L, Zhang A, Wang J, Liu L, Qu C, Li J. Genome-wide analysis and expression profiling of the SUC and SWEET gene families of sucrose transporters in oilseed rape (Brassica napus L.). Front Plant Sci. 2016;7:1464.
Deshmukh RK, Sonah H, Singh NK. Intron gain, a dominant evolutionary process supporting high levels of gene expression in rice. J Plant Biochem Biotechnol. 2016;25:142–6.
Savage DF, Egea PF, Robles-Colmenares Y, O'Connell JD III, Stroud RM. Architecture and selectivity in aquaporins: 2.5 Å X-ray structure of aquaporin Z. PLoS Biol. 2003;1:e72.
Reuscher S, Akiyama M, Mori C, Aoki K, Shibata D, Shiratake K. Genome-wide identification and expression analysis of aquaporins in tomato. PLoS One. 2013;8:e79052.
Bienert GP, Chaumont F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta. 2014;1840:1596–604.
Holm LM, Jahn TP, Møller AL, Schjoerring JK, Ferri D, Klaerke DA, Zeuthen T. NH3 and NH 4+ permeability in aquaporin-expressing Xenopus oocytes. Pflugers Arch. 2005;450:415–28.
Liu L-H, Ludewig U, Gassert B, Frommer WB, von Wirén N. Urea transport by nitrogen-regulated tonoplast intrinsic proteins in Arabidopsis. Plant Physiol. 2003;133:1220–8.
Porcel R, Bustamante A, Ros R, Serrano R, Mulet Salort JM. BvCOLD1: a novel aquaporin from sugar beet (Beta vulgaris L.) involved in boron homeostasis and abiotic stress. Plant Cell Environ. 2018, 41:2844–57.
Mao Z, Sun W. Arabidopsis seed-specific vacuolar aquaporins are involved in maintaining seed longevity under the control of ABSCISIC ACID INSENSITIVE 3. J Exp Bot. 2015;66:4781–94.
Gattolin S, Sorieul M, Frigerio L. Mapping of tonoplast intrinsic proteins in maturing and germinating Arabidopsis seeds reveals dual localization of embryonic TIPs to the tonoplast and plasma membrane. Mol Plant. 2011;4:180–9.
Utsugi S, Shibasaka M, Maekawa M, Katsuhara M. Control of the water transport activity of barley HvTIP3; 1 specifically expressed in seeds. Plant Cell Physiol. 2015;56:1831–40.
Béré E, Lahbib K, Merceron B, Fleurat-Lessard P. Boughanmi NG: α-TIP aquaporin distribution and size tonoplast variation in storage cells of Vicia faba cotyledons at seed maturation and germination stages. J Plant Physiol. 2017;216:145–51.
Devi MJ, Sinclair TR, Jain M, Gallo M. Leaf aquaporin transcript abundance in peanut genotypes diverging in expression of the limited-transpiration trait when subjected to differing vapor pressure deficits and aquaporin inhibitors. Physiol Plant. 2016;156:387–96.
Coskun D, Deshmukh R, Sonah H, Menzies JG, Reynolds O, Ma JF, Kronzucker HJ, Bélanger RR. The controversies of silicon's role in plant biology. New Phytol. 2019;221:67–85.
de Carvalho GAB, Batista JSS, Marcelino-Guimarães FC, do Nascimento LC, Hungria M. Transcriptional analysis of genes involved in nodulation in soybean roots inoculated with Bradyrhizobium japonicum strain CPAC 15. BMC Genomics. 2013;14:153.
El Yahyaoui F, Küster H, Amor BB, Hohnjec N, Pühler A, Becker A, Gouzy J, Vernié T, Gough C, Niebel A. Expression profiling in Medicago truncatula identifies more than 750 genes differentially expressed during nodulation, including many potential regulators of the symbiotic program. Plant Physiol. 2004;136:3159–76.
Bienert GP, Bienert MD, Jahn TP, Boutry M, Chaumont F. Solanaceae XIPs are plasma membrane aquaporins that facilitate the transport of many uncharged substrates. Plant J. 2011;66:306–17.
Willigen CV, Postaire O, Tournaire-Roux C, Boursiac Y, Maurel C. Expression and inhibition of aquaporins in germinating Arabidopsis seeds. Plant Cell Physiol. 2006;47:1241–50.
Novikova GV, Tournaire-Roux C, Sinkevich IA, Lityagina SV, Maurel C, Obroucheva N. Vacuolar biogenesis and aquaporin expression at early germination of broad bean seeds. Plant Physiol Biochem. 2014;82:123–32.
Reinhardt H, Hachez C, Bienert MD, Beebo A, Swarup K, Voß U, Bouhidel K, Frigerio L, Schjoerring JK, Bennett MJ. Tonoplast aquaporins facilitate lateral root emergence. Plant Physiol. 2016; 01635.02015.
Dash S, Cannon EK, Kalberer SR, Farmer AD, Cannon SB. PeanutBase and other bioinformatic resources for peanut. In: Stalker HT, Wilson RF, editors. Peanuts Genetics, Processing, and Utilization. Urbana: AOCS Press; 2016. p. 241–52.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. In: Nucleic Acids Symposium Series; 1999. p. 95–8.
Hu B, Jin J, Guo A-Y, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31:1296–7.
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016;45:D200–3.
Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–9.
Stothard P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques. 2000;28:1102–4.
Kumar S, Nei M, Dudley J, Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9:299–306.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845.
Pellegrini-Calace M, Maiwald T, Thornton JM. PoreWalker: a novel tool for the identification and characterization of channels in transmembrane proteins from their three-dimensional structure. PLoS Comput Biol. 2009;5:e1000440.
Clevenger J, Chu Y, Scheffler B, Ozias-Akins P. A developmental transcriptome map for allotetraploid Arachis hypogaea. Front Plant Sci. 2016;7:1446.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357.
Acknowledgements
Not applicable.
Funding
The project was funded by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC), the Agri-Innovation program Growing Forward 2, SaskCanola and Agriculture and Agri-Food Canada and the Canada Research Chairs Program to RRB. The funding bodies played no role in study design and data collection, analysis, interpretation of data and in writing the manuscript.
Availability of data and materials
All data analyzed in this study are provided in additional files.
Author information
Authors and Affiliations
Contributions
SMS, RD, and HS compiled the data, performed analysis, and wrote first draft of the MS, HS performed expression analysis, RRB planned the study, drew the conclusions and contributed to the writing of the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable; this study has not directly involved humans, animals or plants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional Files
Additional file 1:
Conserved domain analysis of aquaporins identified in Arachis duranensis and Arachis ipaensis using CDD tool from NCBI (DOCX 19 kb)
Additional file 2:
Number of aquaporins identified in Arachis duranensis, Arachis ipaensis and Arachis hypogea genome. (DOCX 13 kb)
Additional file 3:
Transmembrane domains in aquaporins identified in Arachis duranensis and Arachis ipaensis using TMHMM and SOSUI servers (DOCX 22 kb)
Additional file 4:
Predicted tertiary (3D) protein structure of Arachis duranensis and Arachis ipaensis aquaporins (DOCX 7066 kb)
Additional file 5:
Details of predicted sub-cellular location of Arachis duranensis aquaporins identified by using Wolfpsort server (DOCX 16 kb)
Additional file 6:
Amino acid sequences of aquaporins from Arabidopsis thaliana, Glycine max and Oryza sativa used for BLASTp search. Sequence names of A. thaliana, G. max and O. sativa are preceded by the prefixes At, Gm and Os respectively. (DOCX 27 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shivaraj, S.M., Deshmukh, R., Sonah, H. et al. Identification and characterization of aquaporin genes in Arachis duranensis and Arachis ipaensis genomes, the diploid progenitors of peanut. BMC Genomics 20, 222 (2019). https://doi.org/10.1186/s12864-019-5606-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5606-4