
Abstract
Background
Management requires a robust understanding of between- and within-species genetic variability, however such data are still lacking in many species. For example, although multiple population genetics studies of the peregrine falcon (Falco peregrinus) have been conducted, no similar studies have been done of the closely-related prairie falcon (F. mexicanus) and it is unclear how much genetic variation and population structure exists across the species’ range. Furthermore, the phylogenetic relationship of F. mexicanus relative to other falcon species is contested. We utilized a genomics approach (i.e., genome sequencing and assembly followed by single nucleotide polymorphism genotyping) to rapidly address these gaps in knowledge.
Results
We sequenced the genome of a single female prairie falcon and generated a 1.17 Gb (gigabases) draft genome assembly. We generated maximum likelihood phylogenetic trees using complete mitochondrial genomes as well as nuclear protein-coding genes. This process provided evidence that F. mexicanus is an outgroup to the clade that includes the peregrine falcon and members of the subgenus Hierofalco. We annotated > 16,000 genes and almost 600,000 high-quality single nucleotide polymorphisms (SNPs) in the nuclear genome, providing the raw material for a SNP assay design featuring > 140 gene-associated markers and a molecular-sexing marker. We subsequently genotyped ~ 100 individuals from California (including the San Francisco East Bay Area, Pinnacles National Park and the Mojave Desert) and Idaho (Snake River Birds of Prey National Conservation Area). We tested for population structure and found evidence that individuals sampled in California and Idaho represent a single panmictic population.
Conclusions
Our study illustrates how genomic resources can rapidly shed light on genetic variability in understudied species and resolve phylogenetic relationships. Furthermore, we found evidence of a single, randomly mating population of prairie falcons across our sampling locations. Prairie falcons are highly mobile and relatively rare long-distance dispersal events may promote gene flow throughout the range. As such, California’s prairie falcons might be managed as a single population, indicating that management actions undertaken to benefit the species at the local level have the potential to influence the species as a whole.
Similar content being viewed by others


Background
Management of species occurs at multiple scales, requiring a robust understanding of between- and within-species genetic variability. For example, identification of cryptic species (e.g., giraffes [1]) and subspecies (e.g., chimpanzees [2]) allows resources to be allocated to previously unrecognized lineages. Furthermore, an understanding of “evolutionary distinctiveness” (i.e., how isolated a species is within a phylogeny) can result in unique lineages being prioritized for protection [3, 4]. At the population level, an understanding of within-species structure [5,6,7] and adaptive genetic differentiation [8,9,10] allows biologists to identify evolutionarily distinct and/or demographically independent population units of conservation interest [11,12,13] and assign conservation priority.
However, the extent to which genetic variability, population structure and phylogenetic relationships are documented varies drastically across species. For example, multiple population genetics studies of the peregrine falcon (F. peregrinus) have been conducted throughout the species range (e.g., [14,15,16,17,18]) but no similar studies have been done of the closely-related prairie falcon (F. mexicanus). Genomic tools (e.g., whole genome sequencing and SNP genotyping arrays) can rapidly provide insight in species whose genetics have been historically under-studied. High-throughput sequencing and/or SNP assays allow hundreds or thousands of loci to be quickly and affordably genotyped. Larger suites of markers produce more accurate assessments of genome-wide heterozygosity and lead to statistically rigorous phylogenetic reconstructions [19].
Herein, we describe the genomic approaches taken to describe genetic diversity in the prairie falcon (Falco mexicanus) relative to other species and across populations. The prairie falcon range extends from Canada (e.g., British Columbia and Alberta) into the western United States (Washington, Idaho and Montana) south to California, Arizona, New Mexico and ultimately into Mexico [20]. F. mexicanus nests on cliffs and thrives in diverse habitats throughout western North America – from desert and shrub-steppe to grassland and oak-savannah-chaparral [21, 22]. Prairie falcons prefer to feed on ground squirrels even when they are rare relative to other prey species, which include passerines, reptiles, insects and other small mammals [21, 23].
F. mexicanus populations can be adversely affected by anthropogenic development [24,25,26]. Humans indirectly affect prairie falcons by altering natural habitats and decreasing the availability of prey, foraging opportunities or nesting sites. For example, Steenhof et al. [27] argued that spatial patterns of abundance and productivity stemmed from decreased foraging opportunities likely associated with interactions among military training activities, fire and grazing intensity in the Morley Nelson Snake River Birds of Prey National Conservation Area in Idaho. Collisions with wind turbines, in turn, represent a direct threat to prairie falcons [28]. Across the F. mexicanus range, population numbers as indicated by migration data and Western Breeding Bird Survey data appear stable or increasing [29]. However, Christmas Bird Counts decreased linearly between 1977 and 2001 ([29], but see [30]) and declines of occupied nesting territories have been noted locally (e.g., San Francisco East Bay Area; unpublished observations, DA Bell).
An understanding of the underlying genetic variation present in western F. mexicanus is integral to managing the species, as variability is a requirement for species to respond to changing environments and selection pressures [31,32,33]. Furthermore, it is unclear whether prairie falcons in the western United States represent a randomly mating population or genetically distinct units that should be managed separately. To evaluate the current status of the prairie falcon, we developed a draft genome sequence and SNP assay, with the aim of better understanding genetic variability, population structure and adaptive genetic differentiation throughout California and Idaho. Of particular interest is the extent to which gene flow exists amongst prairie falcons nesting in three separate geographic regions in California: the San Francisco East Bay Area, Pinnacles National Park and the Mojave Desert. These areas are undergoing rapid development or are subject to extensive land-use changes, potentially threatening local nesting F. mexicanus.
In addition to this work, we take advantage of our sequencing approach to explore the phylogenetic relationship of the prairie falcon to other falcon species. Historically, the prairie falcon was clustered into the subgenus Hierofalco, which includes the lanner falcon (F. biarmicus), saker falcon (F. cherrug), lager falcon (F. jugger) and gyrfalcon (F. rusticolus), based on ecological and morphological similarities [34, 35]. Subsequent phylogenies generated from sequencing data have indicated that F. mexicanus is more closely related to F. peregrinus than to the hierofalcons. However, branching patterns differ amongst these phylogenies which are based on relatively small portions of the mitochondrial genome [36,37,38,39]. Accordingly, we use nuclear protein-coding genes and the complete mitochondrial DNA sequence of the prairie falcon, described herein, to revisit the phylogeny of Falco.
Methods
Nuclear genome sequencing, assembly and annotation
A female prairie falcon was captured in Siskiyou County, California on 7 June 2014. Two drops of blood were collected via venipuncture of the brachial vein and preserved in lysis buffer (100 mM tris hydrochloric acid, 100 mM ethylenediaminetetraacetic acid, 10 mM sodium chloride, 2% sodium dodecyl sulfate). We extracted DNA (deoxyribonucleic acid) using potassium acetate extraction [40].
We conducted one lane each of paired-end (PE; read length: 100 bp [base pairs]; average fragment length: 568 bp) and mate-paired (MP; read length: 100 bp; average fragment length: 2210 bp) sequencing using an Illumina HiSeq2000 (Table 1). Trimmomatic [41] was used to remove adaptors, discard short reads (< 30 bp), and trim poor quality bases (Illumina Q-value ≤20) from both 5′ and 3′ ends of raw sequence reads. The process described above is appropriate given that the program used for genome assembly accounts for the presence of low quality nucleotides and overly stringent trimming decreases assembly quality [42]. Similarly, GATK (the pipeline used for SNP discovery, see below) requires only the removal of adaptor sequences and subsequently addresses sequencing errors and duplicate reads internally [43, 44]. Fragment lengths and insert sizes were estimated using Picard (http://broadinstitute.github.io/picard).
We additionally generated Illumina TruSeq Synthetic Long Reads (LRs; [45, 46]). To complete the LR sequencing process, we 1) selected 384 genomic DNA fragments 10 kb (kilobases) in length, each of which underwent additional fragmentation, tagging and indexing in an individual well, 2) pooled and purified genomic material from all 384 wells and 3) sequenced the libraries on a single lane using an Illumina HiSeq2000. We again removed adaptors, discarded short reads and trimmed poor quality bases (see above) from the 100 bp reads and the program SPAdes 3.1.1 [47] was used to assemble sequenced fragments into ~ 10 kb LRs.
We used ABySS 1.5.2 [48] to conduct several preliminary assemblies of PE and LR reads, using kmer lengths ranging from 35 to 90. We determined that kmer lengths of 50 or 60 produced the best assemblies by considering both N50 values and the length of the longest scaffold. Final draft assemblies were completed by assembling PE reads into contigs before using both LR and MP reads in the scaffolding step, considering kmer lengths of just 50 and 60. The best draft assembly was chosen by considering both N50 values and the length of the longest scaffold. CEGMA 2.5 [49] was used to identify core eukaryotic genes present in the draft assembly.
We used the MAKER 2.28 pipeline to annotate the draft prairie falcon genome as in Doyle et al. [50]. Briefly, RepeatMasker [51] identified and masked stretches of repetitive DNA, while SNAP [52] and AUGUSTUS [53] were used to generate ab initio gene predictions. Gene predictions were subsequently elevated to gene annotations if expressed sequence tag (EST), protein or InterProScan evidence supported the prediction. Falco cherrug EST sequences were assembled using Trinity as described in Doyle et al. [50]. Gallus gallus, Meleagris gallopavo, Taeniopygia guttata and Columba livia protein sequences were downloaded from the UniProtKB database. InterProScan 5.14 was additionally used to assign gene ontologies to all annotations.
Mitochondrial genome assembly, annotation and phylogenetic analyses
We used baiting and iterative mapping in MITObim 1.6 [54] to create an initial draft of the mitochondrial genome, using a F. mexicanus COI (cytochrome c oxidase subunit I) barcode sequence (AY666553) to initiate assembly. As a quality control measure, we identified mitochondrial sequence reads by blasting to the peregrine falcon mitochondrial genome (AF090338) and subsequently assembled these reads de novo into 38 high-quality contigs using Sequencer 5.4.6. These high-quality contigs were aligned to the MITObim assembly using Sequencer and any disagreements were resolved by eye. The final mitochondrial genome sequence was annotated using MITOS [55].
To generate a phylogenetic tree we used our F. mexicanus mitochondrial genome assembly and all Falco mitochondrial genome sequences available from NCBI (F. peregrinus, AF090338; F. rusticolus, KT989235; F. cherrug, KP337902; merlin, F. columbarius, KM264304; American kestrel, F. sparverius, DQ780880; common kestrel, F. tinnunculus, EU196361; lesser kestrel, F. naumanni, KM251414) and an outgroup (striated caracara, Phalcoboenus australis, KP064202). The latter species was chosen as an outgroup because it was the most complete and closely related mitochondrial genome available that was not of the genus Falco. We used CLUSTALW implemented by MEGA 7.0.21 [56] to align sequences. This alignment was used to produce a maximum likelihood tree using the GTR + G model of evolution and 1000 bootstraps.
Phylogenetic analysis of orthologous genes
We additionally generated a phylogenetic tree using protein sequences from the three available falcon genomes (F. cherrug, F. peregrinus [57]; F. mexicanus, this study). For context, we additionally included sequences from all avian species available through Ensembl (Gallus gallus, Meleagris gallopavo, Anas platyrhynchos, Ficedula albicollis and Taeniopygia guttata [58]) as well as an outgroup (Anolis carolinensis [58]). Orthologous gene families were identified using BLAST® 2.3.0 and OrthoMCL 2.0.9 [59, 60]. Single-copy orthologs present in all species were extracted using custom bash scripts and aligned using MUSCLE 3.8.31 [61]. We subsequently trimmed the alignment using trimAl [62] and generated a super matrix with FASConCAT [63]. We used RAxML [64] to generate a maximum likelihood tree using the JTT + I + G + F model of evolution and 1000 bootstraps.
SNP genotyping
We aligned the PE sequence reads back to the draft prairie falcon genome assembly using BWA [65]. We then used Picard (http://broadinstitute.github.io/picard) to sort mapped reads and identify duplicates. We used GATK 3.2 [43, 44] to identify and realign reads around insertions/deletions (indels) and subsequently call high-quality SNPs (Phred quality score ≥ 30, no more than two alleles for nuclear SNPs and a minimum depth of 10 reads) while masking indels.
We used SnpEff [66] to identify nuclear SNPs present in exonic regions, as well as predict the effects of variants on genes (i.e., amino acid changes). SNPs present in the exons of genes were annotated using BLAST® 2.2.3. We used IGV 2.3 [67, 68] to identify target SNPs with at least 60 nucleotides of high-quality flanking sequence upstream and downstream, GC content less than 65%, and no other variable sites within 20 nucleotides. We deliberately minimized linkage disequilibrium by choosing a single SNP from each annotated gene. Ultimately, we developed 190 autosomal nuclear markers from protein-coding genes. Half (95) of the gene-associated markers were specifically targeted because of evidence for selection in other species (Additional file 1: Table S1). For the remaining 95 gene-associated markers, we preferentially chose SNPs with nonsynonymous amino acid changes to increase the likelihood of identifying genes under selection, as such genes can be early indicators of population differentiation [69,70,71,72]. We additionally identified two molecular sexing markers, each of which represents a single nucleotide difference between the Z- and W-chromosomes of the CHD1 gene. All 192 markers were incorporated into a Fluidigm® SNP Type™ assay.
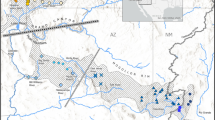
We genotyped 103 individual prairie falcons using the Fluidigm® BioMark HD™ Genotyping System. Blood samples were taken from 89 individuals in California and preserved in Longmire’s lysis buffer [73]. Blood samples were opportunistically collected from 14 individuals in Snake River Birds of Prey National Conservation Area in Idaho during a study of long-range movements [74]. Following sample collection, each individual prairie falcon was released. Of the 89 California individuals, 37 were sampled in and immediately around the San Francisco East Bay Area, 32 from Pinnacles National Park, 17 in the Mojave Desert and three from Northern California (Fig.1). Individuals sampled in both California and Idaho included chicks, juveniles and adults (Table 2). DNA extraction was performed using ammonium acetate [75] and potassium acetate extraction [40].

Sampling locations from San Francisco East Bay Area (CA), Pinnacles National Park (CA), the Mojave Desert (CA), Northern California and Snake River Birds of Prey National Conservation Area (ID). The map layer came from National Geographic, the breeding range layer from Birds of North America Online (https://birdsna.org), maintained by the Cornell Lab of Ornithology [20]
To assess the repeatability of the assay, two additional replicates from nine individuals were also included, for a total of 121 F. mexicanus samples. We subsequently edited individual SNP calls using the Fluidigm® Genotyping Analysis Software. Markers were excluded from downstream analyses if: 1) data did not cluster into distinct homozygous and heterozygous states, 2) minor allele frequencies were less than 0.025 or 3) there was evidence of linkage disequilibrium (i.e., D’ > 0.20) associated with two markers, in which case only one of the two markers was removed. We calculated allele frequencies and linkage disequilibrium using the programs GenAlEx 6.501 [76] and snpStats [77].
Following Doyle et al. [5], we quantified error rates associated with SNP genotyping using three replicate samples from 9 individuals (27 samples in total). We used GenAlEx 6.501 to estimate the probability of identity (PID). PID quantifies the probability that two randomly chosen individuals in a population will have identical genotypes [78] and thus indicates whether a genotyping assay can be used to assign opportunistically collected samples (e.g., feathers) to individuals. To test the accuracy of our molecular sexing approach, we determined the sex of a subset of 67 individuals using our novel markers (hereafter referred to as CHD1_1 and CHD1_2) as well as a traditional PCR (polymerase chain reaction)/gel method [79].
Genetic variation and population structure
GenAlEx 6.501 [76] was used to calculate observed and expected heterozygosity (HO and HE) as well as determine which loci were out of Hardy-Weinberg Equilibrium and exhibited heterozygote excess and deficiency. We tested the null hypothesis that the prairie falcons sampled are part of a single panmictic population using a combination of approaches. First, we conducted a Bayesian analysis with STRUCTURE 2.3.4 [80] and Structure Harvester [81]. Included in the analysis were 54 chicks sampled in California (i.e., individuals that have not yet had the opportunity to disperse and as such represent known-provenance birds). We subsequently conducted an additional test of panmixia using STRUCTURE 2.3.4 and 90 genotypes from both chicks and adults sampled in California and Idaho. This represents a less conservative approach (as adults may have dispersed prior to sampling) but allows us to consider population structure across a larger portion of the prairie falcon range. In both analyses, we retained only one family member genotype whenever family members were known (i.e., parent and chick or siblings) to prevent clustering algorithms from confusing family groups for population structure [82]. The 20 loci not in Hardy-Weinberg equilibrium were excluded. We considered values of K = 1–8, running each value 10 times with an initial burn-in of 100,000 MCMC (Markov chain Monte Carlo iterations) and 1,000,000 subsequent iterations for each value. We assumed an admixture ancestry model and allowed for correlated allele frequencies [83]. The results of both analyses were interpreted using mean likelihood values of K and ΔK [84]. Second, we calculated locus-specific and global pairwise FST (fixation index) values for individuals sampled in the geographically distinct regions of the San Francisco East Bay Area, Pinnacles National Park, the Mojave Desert and Snake River Birds of Prey National Conservation Area using diveRsity [85].
We used two approaches to investigate whether locus-specific signatures of natural selection were present. LOSITAN [86] was run with 500,000 replicates assuming an infinite alleles mutation model. We tested for outliers assuming a confidence interval of 0.99 and a false discovery rate (FDR) rate of 0.05. BAYESCAN [87] was initialized with 10 pilot runs of 5000 iterations and an additional burn-in of 50,000 iterations. We subsequently used a total number of 150,000 iterations (samples size of 5000 with a thinning factor of 20) to identify outlier loci by FST amongst the geographically distinct regions of the San Francisco East Bay Area, Pinnacles National Park, the Mojave Desert and Snake River Birds of Prey National Conservation Area.
Results
Mitochondrial and nuclear genome assembly and annotation
We generated 127 Gb of raw sequence data from F. mexicanus, including 41.1 Gb from the PE library, 35.0 Gb from the MP library and 50.9 Gb from the LR library (Table 1). LR fragments were assembled to form 384 LR reads. Our draft nuclear genome assembly includes 4660 scaffolds greater than 2000 bp (Table 3). These scaffolds had an N50 of 3713 kb and the longest scaffold was 17,400 kb in length. CEGMA indicated that 89% of core eukaryotic proteins were present in the draft assembly.
We annotated 2181 scaffolds greater than 10 kb (N50: 3718), as shorter scaffolds rarely produce high-quality gene annotations and greatly increase computation time (C. Holt, personal communication). The PE coverage of these 2181 scaffolds (which is most relevant because only PE reads were subsequently used for SNP discovery, see below) was approximately 31X (Additional file 2: Figure S1). This process produced 16,320 gene annotations (Table 3). Mean gene length was 16,289 and on average, 9.9 exons were predicted in each gene. Mean exon and intron lengths were 148 and 2470 bp, respectively. Gene ontologies were assigned to 89% of the F. mexicanus genes and the top 100 protein domains can be found in Additional file 3: Table S2.
The F. mexicanus mitochondrial genome assembly was 17,117 bp in length and characterized by 13 protein-coding genes, two ribosomal subunit genes, 22 transfer RNA genes and a control region (Additional file 4: Figure S2). The assembled mitochondrial genome was approximately 1000 bp shorter than that of F. peregrinus and the hierofalcons, which can be largely attributed to a shorter pseudo-control region in F. mexicanus. As in many falcon species, the prairie falcon pseudo-control region was largely dominated by a repetitive region [36, 88]. As such, F. mexicanus may truly have a shorter pseudo-control region, as do the kestrels (e.g., F. tinnunculus and F. naumanni), or a longer repetitive region may have been collapsed during assembly. The assembly was ~ 94% identical to that of the F. rusticulus, F. peregrinus and F. cherrug mitochondrial genome sequences.
Mitochondrial and nuclear phylogenetic analyses
Our maximum likelihood phylogenetic tree generated from complete mitochondrial genome sequences indicates that F. mexicanus is an outgroup to the clade that includes F. peregrinus and the hierofalcons (i.e., F. rusticulus and F. cherrug), with 100% bootstrap support for the relevant branching patterns (Fig. 2a). Our OrthoMCL analysis identified 3770 single-copy orthologs present in all 9 species. Broader phylogenetics relationships among avian species echoed those of recent publications (e.g., the chicken, turkey and duck form an evolutionary branch distinct from that of the falcons, zebra finch and collared flycatcher [57, 89, 90]; Fig. 2b). The maximum likelihood phylogenetic tree generated from nuclear protein-coding sequences again indicates that F. mexicanus is an outgroup to the clade that includes F. peregrinus and the hierofalcon F. cherrug (Fig. 2b).

a A phylogeny of falcon species and an outgroup (P. australis) built using complete mtDNA genome sequences. A CLUSTALW alignment was used to produce a maximum likelihood tree with the GTR + G model of evolution and 1000 bootstraps. Bootstrap values < 50% are not shown on the tree. b A phylogeny of F. peregrinus, F. cherrug, F. mexicanus, G. gallus, M. gallopavo, A. platyrhynchos, F. albicollis and T. guttata and an outgroup (A. carolinensis) built using 3770 single-copy orthologs. A MUSCLE alignment was used to produce a maximum likelihood tree with the JFF + I + G + F model of evolution and 1000 bootstraps
SNP assay development and genotyping
We initially identified 567,599 high-quality SNPs. Of these, 7401 were found in the exons of genes. As described in the methods, the 190 autosomal nuclear markers subsequently included in our SNP assay were chosen for their high-quality flanking sequence, to minimize linkage disequilibrium and maximize the likelihood of identifying genes under selection. Following genotyping, we excluded from downstream analysis 47 loci for reasons outlined in the methods (e.g., minor allele frequencies less than 0.025). Of the remaining 143 loci used to generate all results described below, at least 133 loci amplified for each of the 103 prairie falcons genotyped.
Our error rate, calculated following Doyle et al. [5] and based on three replicate samples taken from each of 9 individuals, was 0.3%. PID was estimated as 1.1 × 10− 43. Our CHD1_1 and CHD1_2 sexing markers were 92 and 100% concordant with Fridolfsson and Ellegren’s [79] PCR/gel molecular sexing method, respectively. All instances of disagreement between CHD1_1 and other molecular sexing methods indicated allelic dropout (i.e., females misidentified as males). CHD1_2 was therefore used for all subsequent molecular sexing. Of the 103 prairie falcons genotyped, 53 were female and 50 male (Table 2).
Heterozygosity and population structure
Mean HO and HE at autosomal SNPs were both 0.34 ± 0.01 SE. Of the 143 nuclear loci considered, 20 were out of Hardy-Weinberg Equilibrium. FIS (inbreeding coefficient) values for these 20 SNPs ranged from − 0.31 to 0.50, with 14 markers showing evidence of heterozygote deficiency and 6 showing evidence of heterozygote excess. When samples from California and Idaho are considered separately, average HO and HE varied from 0.33 ± 0.01 SE to 0.35 ± 0.02 SE and 0.32 ± 0.01 SE to 0.34 ± 0.01 SE, respectively (Table 2).
Both STRUCTURE analyses (i.e., conservative and relaxed approaches) provide evidence that individual prairie falcons in California and Idaho make up a single, panmictic population (Fig. 3a and b, Additional file 5: Figure S3). Mean likelihood values of K are greatest for K = 1 in both instances. Global pairwise FST values for four putative populations (i.e., the San Francisco East Bay Area, Pinnacles National Park, the Mojave Desert and Snake River Birds of Prey National Conservation Area) ranged from 0.01 to 0.03 and did not indicate significant genetic differentiation (Table 4). Our LOSITAN analysis identified two outlier SNPs potentially under directional selection and associated with genes CACNA1G and A2ML1 (Additional file 6:Figure S4). BAYESCAN did not detect any statistically significant outlier loci, however the SNP associated with A2ML1 showed clear differentiation from other markers (Additional file 6: Figure S4). Locus-specific pairwise FST comparisons for A2ML1 indicate high levels of genetic differentiation (i.e., FST > 0.10) between the San Francisco East Bay Area and Idaho, the San Francisco East Bay Area and the Mojave Desert, Idaho and Pinnacles National Park and the Mojave Desert and Pinnacles National Park (Additional file 7: Table S3).

STRUCTURE results consistent with panmixia (i.e., K=1) for known and unknown-provenance falcons. a Results of STRUCTURE analysis for 54 known-provenance chicks sampled from California’s San Francisco East Bay Area, Pinnacles National Park and the Mojave Desert that were genotyped at 123 SNP loci. STRUCTURE results were CLUMPP-averaged across 10 runs when K is assumed to be equal to two. b Results of STRUCTURE analysis for a mix of 90 known and unknown provenance individuals sampled in California and Idaho and genotyped at 123 SNP loci. STRUCTURE results were CLUMPP-averaged across 10 runs when K is assumed to be equal to two
Discussion
Nuclear and mitochondrial genome assembly, annotation and phylogenetics
Herein, we describe the draft genome assembly of F. mexicanus, a species for which population-level genetic variability is undocumented and phylogenetic relationships to other falcons contested. The assembly size (1.17 Gb) and the number of genes annotated (> 16,000) are very similar to that of the F. peregrinus and F. cherrug genomes ([57]; Table 3). The overall completeness of the genome is further indicated by the number of core eukaryotic genes identified (89%), which is comparable to other high quality avian genome assemblies (e.g., rock pigeon [91]).
Our maximum likelihood phylogenetic trees utilizing complete mitochondrial genome sequences and nuclear protein-coding sequences position F. mexicanus as an outgroup to the clade that includes F. peregrinus and the hierofalcons (as in [39]) rather than as a sister species to F. peregrinus (as in [36, 37]). As such, the ecological and morphological similarities between F. mexicanus and the hierofalcons (e.g., syringeal characters [92]) might simply be conserved characters present in many falcon species, rather than evidence of a close evolutionary relationship. It should be reiterated, however, that in our nuclear phylogeny the hierofalcons are represented by a single species (F. cherrug) and additional sequencing will pave the way for fine-scale resolution of branching patterns within Falconinae as well as Neoaves as a whole. For example, an orthologous gene set of protein-coding genes, introns and nonoverlapping ultraconserved elements illustrated that falcons, although traditionally grouped with other diurnal raptors, are more closely related to seriemas, parrots and members of Passeriformes ([90], see also [89]). More accurate estimates of branch lengths, in turn, can improve our estimates of evolutionary distinctiveness, allowing conservation priority to be assigned to species based not just on conservation status (e.g., IUCN rankings) but also by how much evolutionary information would be lost if the species became extinct.
SNP assay development and genotyping
Common molecular approaches (e.g., genotyping with a species-specific suite of microsatellite markers) have been underutilized in F. mexicanus. As a result, little is known about the population genetics of the species throughout its range. Our novel SNP assay is a powerful tool in addressing gaps in our understanding. As with assays designed for golden eagles [5] and grey whales [93], SNP genotyping produced both a low error rate and PID (probability of identity). A low PID indicates that, for example, two naturally shed feathers with identical genotypes were likely derived from the same individual and could be so assigned. As a result, our approach can be applied to noninvasive sampling in addition to the genotyping of high-quality samples taken from known individuals (as practiced in this study). Noninvasive sampling and subsequent DNA extraction from naturally shed hair, feathers, fecal matter and carcasses has facilitated studies of dispersal (wolves, Canis lupis [94]), population size (brown bears, Ursus arctos [95]), sex ratio (Eurasian otter, Lutra lutra [96]), movement (white-tailed eagles, Haliaeetus albicilla [97]), mating systems, population turnover and behavior (imperial eagles, Aquila heliaca [76, 98]).
Additionally, our assay incorporates a molecular sexing marker that is in complete accordance with traditional molecular sexing methods. Finally, the incorporation of ~ 140 gene-associated SNPs has a number of potential benefits. For example, heterozygosity estimated from a large suite of SNPs may reflect genome-wide genetic variation more accurately than other methods (e.g., microsatellites [99]), facilitating future studies of heterozygosity-fitness correlations.
Genetic variation and population structure
We tested the null hypothesis that prairie falcons in the western United States make up a single, interbreeding population, as well as the alternative hypothesis that genetically distinct populations exist. There are biological arguments for each scenario. Most avian species are highly mobile, capable of long-distance movement and able to surmount landscape features that act as barriers to other species (e.g., mountain ranges, rivers), promoting gene flow. As a result, species such as mallards (Anas platyrhynchos) and turtle doves (Streptopelia turtur) exhibit little to no population structure even at a continental level [100, 101]. However, mobility does not necessarily indicate dispersal to and inclusion in novel breeding populations. Avian species can also exhibit natal philopatry and site fidelity that interrupts gene flow and contributes to population structure (e.g., black-browed albatrosses, Thalassarche melanophris [102]; saltmarsh sparrows, Ammodramus caudacutus [103]; penguins, Pygoscelis papua [104]; white-tailed sea eagles, Haliaeetus albicilla [105]).
Banding and telemetry data gives us an indication of F. mexicanus mobility and dispersal. Prairie falcons breeding in Canada and Idaho are known to migrate up to 1900 and 4600 km (kilometers), respectively [74, 106, 107], indicating an ability to travel long distances. However, nestlings banded at Snake River Birds of Prey National Conservation Area have a relatively conservative mean dispersal distance from natal to breeding territories of ~ 9 km [108]. Adult prairie falcons also show a tendency toward breeding territory fidelity. For example, telemetry data indicates that most adult prairie falcons studied at Snake River Birds of Prey National Conservation Area are loyal to their nesting sites across years (i.e., return to within 2.5 km of the previous year’s nesting site; Steenhof et al. [74]). However, exceptions occur. For example, Steenhof et al. [74] documented one of 24 telemetered prairie falcons dispersing between breeding locations 124 km from one another across two years. Relatively few dispersing individuals are required to genetically homogenize populations [109, 110], so even this low level of long-distance movement between breeding locations may be enough to result in a genetically panmictic population. This likely explains the lack of population structure we see throughout California. Additional sampling, however, will be required to determine whether the lack of structure we see between California and Idaho is indicative of the entire western prairie falcon range.
Despite specifically targeting loci likely to be under selection and identifying 20 loci with departures from Hardy-Weinberg equilibrium, our LOSITAN and BAYESCAN analyses identified just two potential outlier loci (CACNA1G and A2ML1) following FDR correction for multiple testing. We will focus the remainder of our discussion on the SNP associated with A2ML1, given the relatively consistent signals of selection from both LOSITAN and BAYESCAN analyses. For this SNP, pairwise FST values indicate that individuals sampled in the San Francisco East Bay Area and Pinnacles National Park differ genetically from individuals sampled in the Mojave Desert and Idaho. A2ML1 is a gene that encodes for a protein that inhibits proteases and is associated with successful embryonic development in chickens and ducks [111, 112]. Interestingly, A2ML1 is considered a candidate reproductive barrier gene isolating the Italian sparrow (Passer italiae) from its two progenitor species: the house and Spanish sparrows (Passer domesticus and Passer hispaniolensis, respectively). Allele frequencies associated with A2ML1 exhibit steep clines throughout the range of the three sparrow species [113, 114]. Although the majority of our analyses indicate that prairie falcons might be managed as a single population, it is possible that the segregating allele frequencies associated with A2ML1 are an early signal of population divergence, as studies have shown that loci under selection show more structure between populations than neutral loci [70]. However, given our small sample size, additional sampling will be required to confirm these results. Furthermore, incorporating markers with different mutation rates and effective population sizes (e.g., intergenic SNPs, microsatellites, mitochondrial sequences) will further expand our understanding of genetic differentiation in the prairie falcon.
Conclusions
Our study illustrates how genomic resources can rapidly shed light on genetic variability at the species- and population-level in understudied species. Our evidence that the prairie falcon is neither sister taxon to the peregrine falcon nor member of the hierofalcons illustrates how a genomic tool set can resolve phylogenies, ultimately contributing to more accurate estimates of evolutionary distinctiveness. Furthermore, our preliminary results largely demonstrate panmixia in the prairie falcon and imply that management actions undertaken to benefit the species at the local level (e.g., regional or park level) have the potential to influence the species as a whole. For example, panmixia indicates a putative tendency for F. mexicanus to disperse throughout its range. This may serve to recover populations locally extirpated as a result of development [26], similar to the sources-sink dynamics demonstrated for recovering peregrine falcon populations in California [115, 116] or the recolonization of volcanic islands post-eruption [117]. Lastly, our sequencing of the prairie falcon genome provides the raw data for subsequent studies of repetitive elements, chromosomal organization and many other research avenues.
Abbreviations
- bp:
-
Basepairs
- COI:
-
Cytochrome c oxidase subunit I
- DNA:
-
Deoxyribonucleic acid
- EST:
-
Expressed sequence tag
- FDR:
-
False discovery rate
- FIS :
-
Inbreeding coefficient
- FST :
-
Fixation index
- Gb:
-
Gigabases
- HE :
-
Expected heterozygosity
- HO :
-
Observed heterozygosity
- kb:
-
Kilobases
- km:
-
Kilometers
- LR:
-
Long reads
- MCMC:
-
Markov chain Monte Carlo iterations
- MP:
-
Mate-paired
- PCR:
-
Polymerase chain reaction
- PE:
-
Paired-end
- PID :
-
Probability of identity
- SNP:
-
Single nucleotide polymorphism
References
Fennessy J, Bidon T, Reuss F, Kumar V, Elkan P, Nilsson MA, Vamberger M, Fritz U, Janke A. Multi-locus analyses reveal four giraffe species instead of one. Curr Biol. 2016;26:1–7.
Bowden R, MacFie TS, Myers S, Hellenthal G, Nerrienet E, Bontrop RE, Freeman C, Donnelly P, Mundy NI. Genomic tools for evolution and conservation in the chimpanzee: Pan troglodytes ellioti is a genetically distinct population. PLoS Genet. 2012;8:1–10.
Redding DW, Mooers AØ. Incorporating evolutionary measures into conservation prioritization. Conserv Biol. 2006;20:1670–8.
Jetz W, Thomas GH, Joy JB, Redding DW, Hartmann K, Mooers AO, Sheffield S. Global distribution and conservation of evolutionary distinctness in birds. Curr Biol. 2014;24:919–30.
Doyle JM, Katzner TE, Roemer GW, Cain JW, Millsap BA, McIntyre CL, Sonsthagen SA, Fernandez NB, Wheeler M, Bulut Z, Bloom PH, DeWoody JA. Genetic structure and viability selection in the golden eagle (Aquila chrysaetos), a vagile raptor with a Holarctic distribution. Conserv Genet. 2016;17:1307–22.
Ruegg KC, Anderson EC, Paxton KL, Apkenas V, Lao S, Siegel RB, DeSante DF, Moore F, Smith TB. Mapping migration in a songbird using high-resolution genetic markers. Mol Ecol. 2014;23:5726–39.
Malenfant RM, Coltman DW, Davis CS. Design of a 9K illumina BeadChip for polar bears (Ursus maritimus) from RAD and transcriptome sequencing. Mol Ecol Resour. 2015;15:587–600.
Zhao S, Zheng P, Dong S, Zhan X, Wu Q, Guo X, Hu Y, He W, Zhang S, Fan W, Zhu L, Li D, Zhang X, Chen Q, Zhang H, Zhang Z, Jin X, Zhang J, Yang H, Wang JJ, Wang JJ, Wei F. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet. 2012;45:67–71.
Schweizer RM, Robinson J, Harrigan R, Silva P, Galverni M, Musiani M, Green RE, Novembre J, Wayne RK. Targeted capture and resequencing of 1040 genes reveal environmentally driven functional variation in grey wolves. Mol Ecol. 2016;25:357–79.
Funk WC, Lovich RE, Hohenlohe PA, Hofman CA, Morrison SA, Sillett TS, Ghalambor CK, Maldonado JE, Rick TC, Day MD, Polato NR, Fitzpatrick SW, Coonan TJ, Crooks KR, Dillon A, Garcelon DK, King JL, Boser CL, Gould N, Andelt WF. Adaptive divergence despite strong genetic drift: Genomic analysis of the evolutionary mechanisms causing genetic differentiation in the island fox (Urocyon littoralis). Mol Ecol. 2016;25:2176–94.
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW. Harnessing genomics for delineating conservation units. Trends Ecol Evol. 2012;27:489–96.
Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, Veeramah KR, Woerner AE, O’Connor TD, Santpere G, Cagan A, Theunert C, Casals F, Laayouni H, Munch K, Hobolth A, Halager AE, Malig M, Hernandez-Rodriguez J, Hernando-Herraez I, Prüfer K, Pybus M, Johnstone L, Lachmann M, Alkan C, Twigg D, Petit N, Baker C, Hormozdiari F, Fernandez-Callejo M, et al. Great ape genetic diversity and population history. Nature. 2013;499:471–5.
Peters JL, Lavretsky P, DaCosta JM, Bielefeld RR, Feddersen JC, Sorenson MD. Population genomic data delineate conservation units in mottled ducks (Anas fulvigula). Biol Conserv. 2016;203:272–81.
Nesje M, Roed K, Bell D, Lindberg P, Lifjeld J. Microsatellite analysis of population structure and genetic variability in peregrine falcons (Falco peregrinus). Anim Conserv. 2000;3:267–75.
Nesje M, Roed K, Lifjeld J, Lindberg P, Steens O. Genetic relationships in the peregrine falcon (Falco peregrinus) analysed by microsatellite DNA markers. Mol Ecol. 2000;9:53–60.
Brown J, van Coeverden de Groot PJ, Birt TP, Seutin G, Boag P, Friesen V. Appraisal of the consequences of the DDT-induced bottleneck on the level and geographic distribution of neutral genetic variation in Canadian peregrine falcons, Falco peregrinus. Mol Ecol. 2007;16:327–43.
Jacobsen F, Nesje M, Bachmann L, Lifjeld JT. Significant genetic admixture after reintroduction of peregrine falcon (Falco peregrinus) in Southern Scandinavia. Conserv Genet. 2008;9:581–91.
Lifjeld JT, Bjornstad G, Steen OF, Nesje M. Reduced genetic variation in Norwegian peregrine falcons Falco peregrinus indicated by minisatellite DNA fingerprinting. Ibis. 2002;144:E19–26.
Oyler-McCance SJ, Oh KP, Langin KM, Aldridge CL. A field ornithologist’s guide to genomics: Practical considerations for ecology and conservation. Auk. 2016;133:626–48.
Steenhof K. Prairie falcon (Falco mexicanus). Birds of North America Online. The Cornell Lab of Ornithology; 2013. https://birdsna.org/Species-Account/bna/species/prafal. Accessed 2 Aug 2017
Sibley D. The Sibley Guide to Birds. 2nd ed. New York: Scott & Nix, Inc; 2014.
Crossley R, Ligouri J, Sullivan B. The Crossley ID Guide: Raptors. Princeton: Princeton University Press; 2013.
Steenhof K, Kochert MN. Dietary responses of three raptor species to changing prey densitites in a natural environment. J Anim Ecol. 1988;57:37–48.
Duerr AE, Miller TA, Cornell Duerr KL, Lanzone MJ, Fesnock A, Katzner TE. Landscape-scale distribution and density of raptor populations wintering in anthropogenic-dominated desert landscapes. Biodivers Conserv. 2015;24:2365–81.
Berry M, Bock C, Haire S. Abundance of diurnal raptors on open space grasslands in an urbanized landscape. Condor. 1998;100:601–8.
Pandolfino E, Herzog M, Hooper S, Smith Z. Winter habitat associations of diurnal raptors in California’s Central Valley. West Birds. 2011;42:62–84.
Steenhof K, Carpenter LB, Kochert MN, Lehman RN. Long-term prairie falcon population changes in relation to prey abundance, weather, land uses, and habitat conditions. Condor. 1999;101:28–41.
Smallwood KS, Thelander C. Bird mortality in the Altamont Pass Wind Resource Area, California. J Wildl Manag. 2008;72:215–23.
Hoffman S, Smith J. Population trends of migratory raptors in Western North America, 1977-2001. Condor. 2003;105:397–419.
Paprocki N, Heath JA, Novak SJ. Regional distribution shifts help explain local changes in wintering raptor abundance: Implications for interpreting population trends. PLoS One. 2014;9
McMahon BJ, Teeling EC, Höglund J. How and why should we implement genomics into conservation? Evol Appl. 2014:1–9.
Frankham R. Stress and adaptation in conservation genetics. J Evol Biol. 2005;18:750–5.
Markert JA, Champlin DM, Gutjahr-Gobell R, Grear JS, Kuhn A, McGreevy TJ, Roth A, Bagley MJ, Nacci DE. Population genetic diversity and fitness in multiple environments. BMC Evol Biol. 2010;10:205.
Cade T. Falcons of the World. London: Collins; 1982.
Kleinschmidt O. Der Formenkreis Hierofalco und die Stellung des ungarischen Wurgfalken in demselben. Aquila. 1901;8:1–48.
Nittinger F, Haring E, Pinsker W, Wink M, Gamauf A. Out of Africa? Phylogenetic relationships between Falco biarmkus and the other Hierofalcons (Aves: Falconidae). J Zool Syst Evol Res. 2005;43:321–31.
Helbig AJ, Seibold I, Bednarek W, Gaucher P, Ristow D, Scharlau W, Schmidl D, Wink M. 1994. Phylogenetic relationships among Falcon species (genus Falco) according to DNA sequence variation of the cytochrome b gene. In: Meyburg B-U, Chancellor RC, editors. Raptor Conservation Today. Berlin: World Working Group Birds of Prey and Pica Press; 1994. p. 593-599.
Wink M, Sauer-Gurth H, Ellis D, Kenward R. Phylogenetic relationships in the Hierofalco complex (Saker-, Gyr-, Lanner-, Laggar Falcon). In: Raptors Worldwide; 2004. p. 499–504.
Wink M, Sauer-Gurth H. Phylogenetic relationships in diurnal raptors based on nucleotide sequences of mitochondrial and nuclear marker genes. In: Raptors Worldwide; 2004. p. 483–98.
Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. 3rd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001.
Bolger A, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS One. 2013;8:e85024.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella K V., Altshuler D, Gabriel S, DePristo MA. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. In Current Protocols in Bioinformatics; 2013(SUPL.43); 1–33.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Voskoboynik A, Neff NF, Sahoo D, Newman AM, Pushkarev D, Koh W, Passarelli B, Fan HC, Mantalas GL, Palmeri KJ, Ishizuka KJ, Gissi C, Griggio F, Ben-shlomo R, Corey DM, Penland L, White RA, Weissman IL, Quake SR. The genome sequence of the colonial chordate, Botryllus schlosseri. elife. 2013;2:e00569.
Mccoy RC, Taylor RW, Blauwkamp TA, Kelley JL, Kertesz M, Pushkarev D, Petrov DA. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetititve transposable elements. PLoS One. 2014;9:e106689.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin A, Sirotkin A, Vyahhi N, Tesler G, Alekseyev M, Pevzner P. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J Comput Biol. 2012;19:455–77.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23.
Parra G, Bradnam K, Ning Z, Keane T, Korf I. Assessing the gene space in draft genomes. Nucleic Acids Res. 2009;37:298–7.
Doyle JM, Katzner TE, Bloom PH, Ji Y, Wijayawardena BK, DeWoody JA. The genome sequence of a widespread apex predator, the golden eagle (Aquila chrysaetos). PLoS One. 2014;9:20–2.
Smit A, Hubley R, Green P. RepeatMasker Open-3.0. http://www.repeatmasker.org. Accessed 10 Aug 2015.
Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59.
Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl. 2):ii215–25.
Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads--a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129.
Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9.
Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9.
Zhan X, Pan S, Wang J, Dixon A, He J, Muller MG, Ni P, Hu L, Liu Y, Hou H, Chen Y, Xia J, Luo Q, Xu P, Chen Y, Liao S, Cao C, Gao S, Wang Z, Yue Z, Li G, Yin Y, Fox NC, Wang J, Bruford MW. Peregrine and saker falcon genome sequences provide insights into evolution of a predatory lifestyle. Nat Genet. 2013;45:563–6.
Kersey PJ, Allen JE, Armean I, Boddu S, Bolt BJ, Carvalho-Silva D, Christensen M, Davis P, Falin LJ, Grabmueller C, Humphrey J, Kerhornou A, Khobova J, Aranganathan K, Langridge N, Lowy E, Mcdowall MD, Maheswari U, Nuhn M, Ong CK, Overduin B, Paulini M, Pedro H, Perry E, Spudich G, Tapanari E, Walts B, Williams G, Ruiz MT, Stein J, et al. Ensembl Genomes 2016: more genomes, more complexity. Nucleic Acids Res. 2016;44:574–80.
Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
van Dongen S. Graph clustering by flow simulation. TheNetherlands: Ph.D thesis, University of Utrecht; 2000.
Edgar RC, Drive RM, Valley M. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Kück P, Meusemann K. FASconCAT: Convenient handling of data matrices. Mol Phylogenet Evol. 2010;56:1115–8.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land S, Ruden D, Lu X. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2, iso-3. Fly (Austin). 2012;6:1–13.
Thorvaldsdóttir H, Robinson J, Mesirov J. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Robinson J, Thorvaldsdottir H, Winckler W, Guttman M, Lander E, Getz G, Mesirov J. Integrative Genomics Viewer. Nat Biotechnol. 2011;29:24–6.
Waples R, Gaggiotti O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol. 2006;15:1419–39.
Freamo H, O’Reilly P, Berg P, Sigbjorn L, Boulding E. Outlier SNPs show more genetic structure between two Bay of Fundy metapopulations of Atlantic salmon than do neutral SNPs. Mol Ecol Resour. 2011;11:254–67.
Helyar SJ, Bekkevold D, Taylor MI, Ogden R, Limborg MT. Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol Ecol Resour. 2011;11:123–36.
Bekkevold D, Helyar SJ, Limborg MT, Nielsen EE, Hemmer-Hansen J, Clausen LA, Carvalho GR. Gene-associated markers can assign origin in a weakly structured fish, Atlantic herring. ICES J Mar Sci. 2015;72:1790–801.
Longmire J, Maltbie M, Baker R. Use of “lysis buffer” in DNA isolation and its implication for museum collections. Occas Pap Museum Texas Tech Univ. 1997;163:1–4.
Steenhof K, Fuller MR, Kochert MN, Bates KK. Long-range movements and breeding dispersal of Prairie Falcons from southwest Idaho. Condor. 2005;107:481–96.
Rudnick JA, Katzner TE, Bragin EA, Rhodes OE, Dewoody JA. Using naturally shed feathers for individual identification, genetic parentage analyses, and population monitoring in an endangered Eastern imperial eagle (Aquila heliaca) population from Kazakhstan. Mol Ecol. 2005;14:2959–67.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics. 2012;28:2537–9.
Clayton D: snpStats: SnpMatrix and XSnpMatrix classes and methods. 2014.
Paetkau D, Strobeck C. Microsatellite analysis of genetic variation in black bear populations. Mol Ecol. 1994;3:489–95.
Fridolfsson A-K, Ellegren HA. simple and universal method for molecular sexing of non-ratite birds. J Avian Biol. 1999;30:116–21.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Earl D, Von Holdt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2011;4:359–61.
Anderson EC, Dunham KK. The influence of family groups on inferences made with the program Structure. Mol Ecol Resour. 2008;8:1219–29.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–87.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Keenan K, McGinnity P, Cross T, Crozier W, Prodohl P. DiveRsity: An R package for the estimation of population genetics parameters and their associated errors. Methods Ecol Evol. 2013;4:782–8.
Antao T, Lopes A, Lopes RJ, Beja-pereira A, Luikart G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics. 2008;9:323.
Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics. 2008;180:977–93.
Sveinsdóttir M, Guðmundsdóttir L, Magnússon KP. Complete mitochondrial genome of the gyrfalcon Falco rusticolus (Aves, Falconiformes, Falconidae). Mitochondrial DNA Part A. 2017;28:370–1.
Hackett SJ, Kimball RT, Reddy S, Bowie RCK, Braun EL, Braun MJ, Chojnowski JL, Cox WA, Han K, Harshman J, Huddleston CJ, Marks BD, Miglia KJ, Moore WS, Sheldon FH, Steadman DW, Witt CC, Yuri T. A phylogenomic study of birds reveals their evolutionary history. Science. 2008;320:1763–9.
Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, Ho SYW, Faircloth BC, Nabholz B, Howard JT, Suh A, Weber CC, da Fonseca RR, Li J, Zhang F, Li H, Zhou L, Narula N, Liu L, Ganapathy G, Boussau B, Bayzid MS, Zavidovych V, Subramanian S, Gabaldon T, Capella-Gutierrez S, Huerta-Cepas J, Rekepalli B, Munch K, Schierup M, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–31.
Shapiro MD, Kronenberg Z, Li C, Domyan ET, Pan H, Campbell M, Tan H, Huff CD, Hu H, Vickrey AI, Neilsen SC, Stringham SA, Hu H, Willerslev E, MTP G, Yandell M, Zhang G, Wang J. Genomic diversity and evolution of the head crest in the rock pigeon. Science. 2013;339:1063–7.
Griffiths C. Syringeal morphology and the phylogeny of the Falconidae. Condor. 1994;96:127–40.
DeWoody JA, Fernandez NB, Brüniche-Olsen A, Antonides JD, Doyle JM, San Miguel P, Westerman R, Vertyankin VV, Godard-Codding CAJ, Bickham JW. Characterization of the gray whale (Eschrichtius robustus) genome and a genotyping array based on single-nucleotide polymorphisms in candidate genes. Biol Bull. 2017;232:186–197.
Valière N, Fumagalli L, Gielly L, Miquel C, Lequette B, Poulle M-L, Weber J-M, Arlettaz R, Taberlet P. Long-distance wolf recolonization of France and Switzerland inferred from non-invasive genetic sampling over a period of 10 years. Anim Conserv. 2003;6:83–92.
Solberg KH, Bellemain E, Drageset O-M, Taberlet P, Swenson JE. An evaluation of field and non-invasive genetic methods to estimate brown bear (Ursus arctos) population size. Biol Conserv. 2006;128:158–68.
Dallas JF, Coxon KE, Sykes T, Chanin PRF, Marshall F, Carss DN, Bacon PJ, Piertney SB, Racey PA. Similar estimates of population genetic composition and sex ratio derived from carcasses and faeces of Eurasian otter Lutra lutra. Mol Ecol. 2003;12:275–82.
Bulut Z, Bragin E, DeWoody J, Braham M, Katzner T, Doyle J. Use of noninvasive genetics to assess nest and space use by white-tailed eagles. J Raptor Res. 2016;50:351–62.
Rudnick JA, Katzner TE, Bragin EA, De Woody JA. A non-invasive genetic evaluation of population size, natal philopatry, and roosting behavior of non-breeding eastern imperial eagles (Aquila heliaca) in central Asia. Conserv Genet. 2008;9:667–76.
DeWoody YD, DeWoody JA. On the estimation of genome-wide heterozygosity using molecular markers. J Hered. 2005;96:85–8.
Kraus RHS, Van Hooft P, Megens HJ, Tsvey A, Fokin SY, Ydenberg RC, Prins HHT. Global lack of flyway structure in a cosmopolitan bird revealed by a genome wide survey of single nucleotide polymorphisms. Mol Ecol. 2013;22:41–55.
Calderón L, Campagna L, Wilke T, Lormee H, Eraud C, Dunn JC, Rocha G, Zehtindjiev P, Bakaloudis DE, Metzger B, Cecere JG, Marx M, Quillfeldt P. Genomic evidence of demographic fluctuations and lack of genetic structure across flyways in a long distance migrant, the European turtle dove. BMC Evol Biol. 2016;16:237.
Burg T, Croxall J. Global relationships amongst black-browed and grey-headed albatrosses: analysis of population structure using mitochondrial DNA and microsatellites. Mol Ecol. 2001;10:2647–60.
Walsh J, Kovach AI, Babbitt KJ, Brien KMO. Fine-scale population structure and asymmetrical dispersal in an obligate salt-marsh passerine, the Saltmarsh Sparrow (Ammodramus caudacutus). Auk. 2012;129:247–58.
Ciborowski KL, Levy H, Clucas GV, Rogers AD, Leach AD, Polito MJ, Lynch HJ, Dunn MJ, Hart T. Population structure and phylogeography of the Gentoo Penguin (Pygoscelis papua) across the Scotia Arc. Ecol Evol. 2016;6:1834–53.
Ponnikas S, Kvist L, Ollila T. Genetic structure of an endangered raptor at individual and population levels. Conserv Genet. 2013;14:1135–47.
Steenhof K, Kochert M, Moritsch M. Dispersal and migration of Southwestern Idaho raptors. J F Ornithol. 1984;55:357–68.
Schmutz J, Fyfe R, Banasch U, Armbruster H. Routes and timing of migration of falcons banded in Canada. Wilson Bull. 1991;103:44–58.
Lehman RN, Steenhof K, Carpenter LB, Kochert M. Turnover and dispersal of prairie falcons in southwestern Idaho. J Raptor Res. 2000;34:262–9.
Spieth T. Gene flow and genetic differentiation. Genetics. 1974;78:961–5.
Wright S. Evolution in Mendelian Populations. Genetics. 1931;16:97–159.
Liu Y, He S, Zeng T, Du X, Shen J, Zhao A, Lu L. Transcriptome analysis of the livers of ducklings hatched normally and with assistance. Asian-Australasian J Anim Sci. 2017;30:773–80.
Cordeiro CMM, Hincke MT. Quantitative proteomics analysis of eggshell membrane proteins during chick embryonic development. J Proteome. 2016;130:11–25.
Elgvin TO, Trier CN, Tørresen OK, Hagen IJ, Lien S, Nederbragt AJ, Ravinet M, Jensen H, Sætre G. The genomic mosaicism of hybrid speciation. Sci Adv. 2017;3:e1602996.
Trier CN, Hermansen JS, Sætre G, Bailey RI. Evidence for mito-nuclear and sex-linked reproductive barriers between the hybrid Italian Sparrow and its parent species. PLoS Genet. 2014;10:e1004075.
Wootton J, Bell DA. metapopulation model of the peregrine falcon in California: Viability and managment strategies. Ecol Appl. 1992;2:307–21.
Wootton J, Bell D. Assessing predictions of population viability analysis: Peregrine falcon populations in California. Ecol Appl. 2014;24:1251–7.
Sonsthagen SA, Williams JC, Drew GS, White CM, Sage GK, Talbot SL. Legacy or colonization? Posteruption establishment of peregrine falcons (Falco peregrinus) on a volcanically active subarctic island. Ecol Evol. 2017;7:107–14.
Huang Y, Li Y, Burt DW, Chen H, Zhang Y, Qian W, Kim H, Gan S, Zhao Y, Li J, Yi K, Feng H, Zhu P, Li B, Liu Q, Fairley S, Magor KE, Du Z, Hu X, Goodman L, Tafer H, Vignal A, Lee T, Kim K-W, Sheng Z, An Y, Searle S, Herrero J, Groenen M, RPM C, et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet. 2013;45:776–83.
Antonides J, Ricklefs R, DeWoody J. The genome sequence and insights into the immunogenetics of the bananaquit (Passeriformes: Coereba flaveola). Immunogenetics. 2017;69:175–86.
Hillier L, Miller W, Birney E, HR WW. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716.
Cai Q, Qian X, Lang Y, Luo Y, Xu J, Pan S, Hui Y, Gou C, Cai Y, Hao M, Zhao J, Wang S, Wang Z, Zhang X, He R, Liu J, Luo L, Li Y, Wang J. Genome sequence of ground tit Pseudopodoces humilis and its adaptation to high altitude. Genome Biol. 2013;14:R29.
Acknowledgements
The authors thank S. Alsup, L. Anderson, C. Battistone, J. Barrett, L. Basulto, G. Beeman, P. Beitz, M. Bell, J. Belli, D. Bittner, W. Boarman, S. Bobzien, S. Buranek, S. Chrisman, M. Clark, P. Connolly, E. Cox, C. Danner, J., A. & L. DiDonato, S. Dulava, L. Encinas, J. Garcia, C. Getzlaff, M. Gnekow, J. Golla, M. Hammond, L. Harp, B. Hart, T. Hatfield, K. Herbinson, P. Hopkinson, K. Hunting, P. Johnson, J. Jones, N. Jones, M. Kochert, B. Latta, E. Leveyas, J. Lincer, A. List, M. Lowry, M. Malec, M. Manapsal, E. & J. Mastrelli, C. Meinke, J. Mohlmann, M. Moran, E. Morata, J. Negreann, F. Muzio, C. Otahal, J. Papp, H. Peeters, R. Pericoli, C. Richardson, D. Riensche, L. Riser, S. Robinson, B. Sandstrom, J. Scullen, V. Seideman, L. Serpa, C. Shafer, J. Sheppard, S. and N. Smallwood, J. Smith, S. Snyder, M. Solomon, M. Sproul, K. Steenhof, C. Swolgaard, E. Tarkata, K. Vanesley, P. & S. Walker, E. Walther, K. & K. Wllitner, A. Welch, A. Wells, H. Wilson, J. Woerner, B. Wolff, R. Woodard, S. Wright, and L. Young, for their assistance in fieldwork and collecting F. mexicanus samples and K. Boettcher and C. McKaskey for assistance generating Fig. 1. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Funding
This work was supported by the U.S. Bureau of Land Management (BLM award numbers L12 AC20102, L11PX02237, and L12 AC2010), the Provost’s Office at Purdue University (University Faculty Scholar program), the National Park Service (PINN645–21), XSEDE (BIO170038), the Wayman-McAuliffe Family Fund for Ornithology, Save Mount Diablo, East Bay Regional Park District, Contra Costa County Fish and Wildlife Propagation Fund, Alameda County Birds of Prey Reserve Foundation and California State University, Sacramento. AF with BLM helped design the sampling scheme and prepare the manuscript.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in NCBI’s Short Read Archive (BioSample accession # SAMN08636363, BioProject accession # PRJNA436967, SRA accession # SRP133947) and the Dryad digital repository (https://doi.org/10.5061/dryad.8b0s04t).
Author information
Authors and Affiliations
Contributions
JMD, DAB, AF, TEK and JAD conceived and designed the research. DAB, PHB, GE, LL and TEK secured permits and collected field data and samples. JMD performed all DNA extractions and associated laboratory procedures. PS and RW sequenced and assembled the genome and JMD and KL performed all other bioinformatic analyses. JMD, DAB and JAD led the writing effort and all authors (JMD, DAB, PHB, GE, AF, TEK, LL, KL, PS, RW and JAD) reviewed, revised, commented on and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Sample collection was approved by the Institutional Animal Care and Use Committees (IACUC) of California State University, Sacramento, CA and Boise State University, Boise, ID. We thank the following agencies for research permits: California Dept. of Fish and Wildlife (SC_007313, SC-000221); US Geological Survey, Bird Banding Laboratory (23,599, 20,431); U.S. Fish and Wildlife Service (MB008248–0); National Park Service (PINN-2015-SCI-0006); Bureau of Land Management – California Desert District (6500 CA-063.50); Central Coast Field Office (CA-190-AP17–01), Carrizo Plain National Monument (04–30-17); California Department of Parks and Recreation (17–820-30); Contra Costa Water District (36); San Francisco Public Utilities Commission (01–25-17); East Bay Municipal Utility District (03–25-16); Livermore Area Recreation and Park District (04–24-04); and The Nature Conservancy and many private landowners who granted access to their lands.
Consent for publication
Not applicable.
Competing Interests
The authors declare they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Description of 96 prairie falcon SNPs associated with genes under selection in different species. (PDF 299 kb)
Additional file 2:
Figure S1. Paired-end read coverage of 2181 scaffolds. Sequencing depth is on the x-axis while the y-axis shows the percentage of total bases at a given depth. Reads were aligned to the genome using BWA. (PDF 156 kb)
Additional file 3:
Table S2. Top Pfam domain hits in the F. mexicanus genome and their counts. (PDF 175 kb)
Additional file 4:
Figure S2. The F. mexicanus mitochondrial genome map. COX1, COX2 and COX3 indicate cytochrome oxidase subunits 1–3; CYTB indicates cytochrome b; atp6 and atp8 indicate ATPase subunits 6 and 8; ND1–ND6 indicate NADH dehydrogenase subunits 1–6. Transfer RNA genes are designated by single-letter amino acid codes. (PDF 192 kb)
Additional file 5:
Figure S3. Mean estimated Ln probability of data ± SD for K 1 through 8, averaged across 10 runs, for known and unknown-provenance falcons. A) Results of STRUCTURE analysis for 54 known-provenance chicks sampled from California’s San Francisco East Bay Area, Pinnacles National Park and the Mojave Desert that were genotyped at 123 SNP loci. B) Results of STRUCTURE analysis for a mix of 90 known and unknown provenance individuals sampled in California and Idaho and genotyped at 123 SNP loci. (PDF 268 kb)
Additional file 6:
Figure S4. Results of LOSITAN and BAYESCAN analyses. a) The confidence area for candidate loci under positive selection is shown in red. LOSITAN identified two outlier loci: X1613239_349604 (CACNA1G) and X1613580_160905 (A2ML1). b) No outlier loci were detected via BAYESCAN analysis. Posterior odds are plotted on the x axis and FST index values on the y axis. (PDF 36 kb)
Additional file 7:
Table S3. Locus-specific pairwise FST values for prairie falcons in the San Francisco East Bay Area (East Bay), Pinnacles National Park (Pinnacles), the Mojave Desert (Mojave) and Idaho. (PDF 281 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Doyle, J.M., Bell, D.A., Bloom, P.H. et al. New insights into the phylogenetics and population structure of the prairie falcon (Falco mexicanus). BMC Genomics 19, 233 (2018). https://doi.org/10.1186/s12864-018-4615-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4615-z