
Abstract
Background
The white-rot fungus Phlebia sp. MG-60 shows valuable properties such as high ethanol yield from several lignocellulosic materials, although white-rot fungi commonly degrade woody components to CO2 and H2O. In order to identify genes involved in ethanol production by Phlebia sp. MG-60, we compared genes differentially expressed by the ethanol producing fungus Phlebia sp. MG-60 and the model white-rot fungus Phanerochaete chrysosporium under ethanol fermenting and non-fermenting conditions using next-generation sequencing technologies.
Results
mRNAs from mycelia of Phlebia sp. MG-60 and P. chrysosporium under fermenting and non-fermenting conditions were sequenced using the MiSeq system. To detect differentially expressed genes, expression levels were measured in fragments per kilobase of exon per million mapped reads (FPKM). Differentially expressed genes were annotated using BLAST searches, Gene Ontology classifications, and KEGG pathway analysis. Functional analyses of differentially expressed genes revealed that genes involved in glucose uptake, glycolysis, and ethanol synthesis were widely upregulated in Phlebia sp. MG-60 under fermenting conditions.
Conclusions
In this study, we provided novel transcriptomic information on Phlebia sp. MG-60, and these RNA-seq data were useful in targeting genes involved in ethanol production for future genetic engineering.
Similar content being viewed by others

Background
Most bioethanol production in the world is currently from food crops, which leads to competition with food and feed uses. Advanced second-generation bioethanol is a renewable transportation fuel made from lignocellulosic biomass that does not compete with food or feed [1]. Lignocellulosic biomass is mainly composed of cellulose, hemicellulose, and lignin and is the most abundant material for bioethanol production. Wood-rot basidiomycetes play key roles in the carbon cycle in forest ecosystems through multi-enzyme systems that degrade lignocelluloses [2]. Wood-rot basidiomycetes are considered the most efficient degraders of lignocellulose in nature. Biological delignification by white-rot basidiomycetes, which is a useful pretreatment for enzymatic saccharification of lignocellulosic biomass, is summarized by Moreno et al. [3]. Basidiomycetes are expected to be used for pretreatment in bioethanol production from lignocellulosic materials. Therefore, some researchers have reported use of white-rot fungi in pretreatment for enzymatic saccharification of lignocellulosic biomass [4–6]. Furthermore, several reports have been published about genome projects on Basidiomycota, Agaricomycetes and Polyporales lignocellulose-degrading fungi, and comparative genomic studies recently [7–10].
Several white-rot fungi have been reported to ferment oligosaccharide materials to ethanol. The white-rot fungi Peniophora cinerea and Trametes suaveolens efficiently convert hexoses to ethanol [11], and Trametes hirsuta shows efficient fermentation of starch, wheat bran and rice straw to ethanol without acid or enzymatic hydrolysis [12]. Recently, Okamoto et al. documented that the white-rot fungus Trametes versicolor KT9427 can produce ethanol from starch, cellulose, xylan, wheat bran and rice straw [13].
The white-rot fungus Phlebia sp. MG-60 was selected as a hypersaline-tolerant lignin-degrading fungus from 28 mushrooms and samples of driftwood based on decolorization and delignification abilities after collection from mangrove stands in Okinawa, Japan [14]. Phlebia sp. MG-60 was capable of converting lignocellulose to ethanol directly with high yield [6, 15, 16]. When this fungus was cultured with 20 g L−1 of unbleached hardwood kraft pulp for 168 h, 71.8 % of the theoretical maximum yield of ethanol was observed, and when it was cultured with waste newspaper, 51.5 % of the theoretical maximum yield was observed [16]. The delignification, anaerobic saccharification, and fermentation of oak wood using only Phlebia sp. MG-60, without addition of chemicals or enzymes, has been also reported [16]. Additionally, alkaline pretreated sugarcane bagasse was fermented well directly, without addition of cellulases, by Phlebia sp. MG-60 [6]. Thus, Phlebia sp. MG-60 possesses not only wood degrading ability but also ethanol fermentability. However, the detailed mechanism of fermentation by this fungus remains unknown.
The goal of this study was to characterize the specific genes for ethanol production, and to predict the mechanism behind the high yield of ethanol by Phlebia sp. MG-60. In the present study, we analyzed differential gene expression of the ethanol producing white-rot fungus Phlebia sp. MG-60 and the model white-rot fungus P. chrysosporium, used as the control, under fermenting and non-fermenting conditions by next-generation sequencing. This is the first report of transcriptomic data of the ethanol producing white-rot fungus Phlebia sp. MG-60.
Methods
Strains
Phlebia sp. MG-60 (MKFC40001) and P. chrysosporium ME-446 (ATCC 34541) were used in this study. Both strains were maintained on potato dextrose agar (PDA) slants at 4 °C.
Production of ethanol from glucose
In order to equalize the growth of mycelia, Phlebia sp. MG-60 and P. chrysosporium were incubated on PDA plates at 30 °C for 5 and 3 days, respectively. 10-mm diameter disks were then punched out from the growing edge of the mycelia using a sterile cork borer. Two mycelial disks for each strain were placed into a 100-mL Erlenmeyer flask containing 10 mL basal liquid medium (20 g L−1 glucose, 10 g L−1 yeast extract, 10 g L−1 KH2PO4, 2 g L−1 (NH4)2SO4, and 0.5 g L−1 MgSO4-7H2O, pH 4.5). After sealing the flask with a silicone plug stopper (to ensure semi-aerobic conditions), the culture was statically incubated at 30 °C for 10 days. Cultures were filtered through a 0.2-μm membrane filter, and the filtrate was then separated by high-performance liquid chromatography (HPLC) using a Shodex SH1821 column (8.0 mm × 300 mm, Showa Denko K.K., Tokyo, Japan) at 75 °C with 0.5 mM H2SO4 as the mobile phase at a flow rate of 0.6 mL min−1, and ethanol and glucose concentrations in the cultures were measured using an online refractive index detector. The pH of the culture was also measured by a glass electrode (D-51S, Horiba Ltd., Kyoto, Japan).
Mycelial dry weight
For monitoring the growth of Phlebia sp. MG-60 and P. chrysosporium, mycelial dry weights obtained from liquid culture which described as above were measured. Cultures were filtered through 0.2-μm membrane filter, mycelium and filter were then dried. The mycelial dry weight calculates by (weight of filter + dried residue) – (weight of filter paper).
cDNA library preparation for DNA sequencing
To construct RNA-seq libraries, total RNA was isolated from the mycelia of Phlebia sp. MG-60 after 2 and 9 days of incubation and from mycelia of P. chrysosporium after 3 and 9 days of incubation. Total RNA was first purified from cultured mycelia with three biological replicates obtained from separate cultures using a Qiagen RNeasy Mini Kit (Hilden, Germany). The quality and quantity of each RNA sample were assessed as described previously [17]. Agarose gel electrophoresis and the OD260/OD280 ratio were used for assessing quality of total RNA.
Each extracted total RNA sample was treated with DNase I and repurified using an RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. Purified RNA (1 μg) was used for first-strand cDNA synthesis using an oligo-dT primer and PrimeScript reverse transcriptase (Takara). An equal quantity of 50 ng total RNA was used for PCR. Primer sequences and the expected product sizes are shown in Additional file 1: Table S1. RT-PCR was performed using 1 μg of total RNA with the PrimeScript RT-PCR Kit and the RT-PCR amplified fragments were analyzed by agarose gel electrophoresis. The libraries for strand-specific RNA sequencing were constructed using a SureSelect Strand-Specific RNA Library prep kit (Agilent Technologies) according to the manufacturer's protocol. We generated libraries of each biological replicates, and then libraries derived from same species were pooled together. The two individual sequencing runs for Phlebia sp. MG-60 and P. chrysosporium were performed, respectively. Transcriptome sequencing of paired-end reads (75 bp) was performed by a MiSeq system (Illumina).
De novo assembly and differential expression analysis
The raw reads were processed using cutadapt version 1.8.1 to remove adapter sequences [18], low-quality bases (quality scores <30) and reads shorter than 50 nt. The last 76 bases were trimmed by FASTX-Toolkit [19]. After quality trimming, the high-quality reads were assembled into unigenes by Trinity (version 2.0.6) [20] Oases (version 0.2.08) [21], Trans-ABySS (version 1.5.4) [22] and SOAPdenovo-Trans (version 1.03) [23]. The resulting unigenes was further analyzed using DETONATE (de novo transcriptome RNA-seq assembly with or without the truth evaluation) [24]. In de novo transcriptome assembly by Trinity program, we selected the Jaccard-clip option to reduce the generation of chimeric transcripts. rRNA were excluded from the unigenes by removing sequences matching entries in the SILVA rRNA database by the Megablast program [25]. The genes derived from mitochondria sequences were manually removed from the unigenes using the result of local BLASTX and BLASTN search against nr and nt database. High-quality short reads were mapped to the rRNA-removed unigenes as a reference using Bowtie [26], and then transcript abundance was estimated using RSEM software [27]. To identify differentially expressed genes (DEGs), P-values and fold changes were computed using the edgeR package [28].
Functional annotation
The unigenes were searched against the Swiss-Prot database by a local BLASTX algorithm (E-value cut-off was set at 10e-5) to predict the biological functions [29]. Open reading frames (ORFs) and their protein sequences were predicted from unigenes using TransDecoder, which is included in the Trinity package. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were assigned to unigenes using the BlastKOALA server [19, 30]. Gene ontology (GO) annotation of the transcriptome was performed using InterproScan software version with the “goterms” option [31]. Orthologous protein pairs between Phlebia sp. MG-60 and P. chrysosporium were identified by the FastOrtho program (http://enews.patricbrc.org/fastortho/) [32] as described previously [33], which is a reimplementation of the OrthoMCL program [34]. The longest peptides translated by TransDecoder were used for orthologous protein analysis as representatives of peptides encoded by the Trinity unigenes. Proteins showing one-to-one correspondence between Phlebia sp. MG-60 and P. chrysosporium were regarded as orthologous proteins.
Semi-quantitative RT-PCR and quantitative RT-PCR (qRT-PCR)
To analyse gene expression in Phlebia sp. MG-60, samples were prepared as described in the section Production of ethanol from glucose. The semi-quantitative RT-PCR method was as described in our previous study [35]. All the selected unigenes used in RT-PCR and qRT-PCR evidenced high similarity with known genes with very low E-values (< E-20), and these transcripts were manually confirmed mapping rate of sequencing reads from each sample and sequence errors using the visualized software IGV tools version 2.0 (http://software.broadinstitute.org/software/igv/home). PCR was performed for 28 cycles, with template denaturation at 95 °C for 30 s, primer annealing at 58 °C for 30 s, and DNA extension at 72 °C for 1 min using Ex Taq DNA Polymerase (TaKaRa Bio).
For the quantitative real-time RT-PCR assays, the Roche LightCycler 480 system was used. Each reaction contained 50 ng of first-strand cDNAs, 2 μL PCR primers, 7 μL water, and 10 μL master mix (Roche). Cycling conditions were set as follows: pre-incubation, 95 °C for 10 min; amplification, 45 cycles of 95 °C for 10 s, 60 °C for 10 s, and 7 °C for 10 s. The reference genes actin and hydroxymethylbilane synthase (HMBS) were used to test for sample-to-sample variation. Relative quantitation using the comparative Ct method was calculated as ΔΔCt = (ΔCttarget –ΔCtcontrol) fermenting conditions − (ΔCttarget – ΔCtcontrol) non-fermenting conditions..
Results
Production of ethanol from glucose
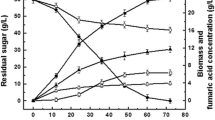
The time courses of ethanol production from glucose and glucose consumption by the ethanol producing white-rot fungus Phlebia sp. MG-60 and the control white-rot fungus P. chrysosporium are shown in Fig. 1. Phlebia sp. MG-60 produced the maximum ethanol concentration of 9.5 g L−1 from glucose after 3 days of incubation, and the concentration then gradually diminished. On the other hand, maximum ethanol production by P. chrysosporium was 2.4 g L−1 after 8 days of incubation (Fig. 1). The glucose concentration of Phlebia sp. MG-60 and P. chrysosporium were 12.8 g L−1 and 18.2 g L−1 at 1 day, and then almost decreased to 0 g L−1 until 2 days and 5 days after incubation, respectively. We also measured the mycelial growth and pH changes in liquid cultures of both species (Additional file 2: Figure S1). Although the growth rate of P. chrysosporium was higher than that of Phlebia sp. MG-60, ethanol productivity of Phlebia sp. MG-60 was much higher than that of P. chrysosporium. The pH in liquid cultures of both species was between 4.5 and 3.8 during the experiment. All the results indicated that there exists a correlation between consumption of glucose and production of ethanol in the mycelia culture of Phlebia sp. MG-60 at 2 days after incubation. The consumed glucose was mainly used for its ethanol production in Phlebia sp. MG-60. The maximum ethanol productivity was at 2 days in Phlebia sp. MG-60 and 3 days of incubation in P. chrysosporium, and that ethanol production stopped at 9 days of incubation in both fungi. Therefore, 2-day-incubated mycelia of Phlebia sp. MG-60 and 3-day-incubated mycelia of P. chrysosporium were used as the fermenting samples, and 9-day-incubated mycelia of Phlebia sp. MG-60 and 9-day-incubated mycelia of P. chrysosporium were used as the non-fermenting samples for further experiments.

Time courses of ethanol production and glucose consumption of Phlebia sp. MG-60 (a) and P. chrysosporium (b). ●: ethanol production; ○: glucose consumption. Values are the means ± SD of triplicate samples
Sequencing and de novo assembly
Total RNA was extracted from Phlebia sp. MG-60 and P. chrysosporium for transcriptome analysis. Samples included biological triplicates of cDNA libraries from Phlebia sp. MG-60 after 2 and 9 days of incubation and P. chrysosporium after 3 and 9 days of incubation (termed MG2D, MG9D, PC3D, and PC9D, respectively). Total mRNA was purified, and then cDNA libraries were constructed and sequenced on the MiSeq platform.
Paired-end sequences (2 x 75 bp in length) from mRNAs of Phlebia sp. MG-60 and P. chrysosporium were generated by MiSeq: 17,822,770 reads (8,911,385 pairs) in MG2D, 25,249,086 reads (12,624,543 pairs) in MG9D, 31,810,504 reads (15,905,252 pairs) in PC3D, and 15,905,252 reads (7,952,626 pairs) in PC9D (Additional file 3: Table S2). After removal of low-quality regions of the reads (quality values <30), each of the high-quality reads from Phlebia sp. MG-60 and P. chrysosporium were assembled into unigenes using the Trinity, Oases, Trans-ABySS and SOAPdenovo-Trans program, and further analyzed using DETONATE. The resulting RSEM-EVAL score from DETONATE analysis indicated that Trinity produced the most accurate assemblies in all data sets. Next, rRNA and genes derived from mitochondria sequences were excluded from Trinity unigenes by removing those matching entries in the SILVA 111 rRNA [36], nr and nt database. Ultimately, we obtained 34,731 (N50 length = 1,422 bp) unigenes in Phlebia sp. MG-60 and 27,956 (N50 length = 1,533 bp) in P. chrysosporium (Table 1).
Functional annotations of differentially expressed genes
All unigenes were searched against the Swiss-Prot database using local BLASTX (Additional file 4: Table S3, Additional file 5: Table S4), and further annotated with GO terms using InterProScan. To identify DEGs, we compared the unigenes that were over 2-fold up- or downregulated with false discovery rate (FDR) < 0.05 between the fermenting and non-fermenting conditions. In Phlebia sp. MG-60, 2,944 genes were upregulated, and 3,910 genes were downregulated (Additional file 4: Table S3). There were 1,689 upregulated genes and 1,901 downregulated genes in P. chrysosporium (Additional file 5: Table S4). We performed GO enrichment analysis of Phlebia sp. MG-60 and P. chrysosporium, which revealed that 27 GO terms were upregulated and 5 were downregulated in Phlebia sp. MG-60. The most enriched GO terms were “ATP binding” (GO: 0005524) in the molecular functions category, “fatty acid biosynthetic process” (GO: 0006633) in biological processes, and “integral component of membrane” (GO: 0016021) in cellular components under fermenting conditions (Table 2). In contrast, only “catalytic activity” (GO: 0003824) in molecular functions was significantly enriched in P. chrysosporium (data not shown).
To evaluate the high ethanol productivity in Phlebia sp. MG-60, we mapped all genes in Phlebia sp. MG-60 and P. chrysosporium to KEGG metabolic pathways using BlastKOALA, and focused on genes involved in ethanol fermentation. Metabolic pathways and biosynthesis of secondary metabolites were the most frequently represented pathways, including glycolysis and pyruvate oxidation, which are involved in ethanol production pathways. Based on the KEGG pathway assignments, we compared the genes involved in the glycolysis/gluconeogenesis pathway in Phlebia sp. MG-60 and P. chrysosporium (Additional file 6: Figure S2). Lists of transcripts related to the glycolysis/gluconeogenesis pathway are shown in Additional file 7: Table S5 and Additional file 8: Table S6. In all, 40 genes were mapped to the glycolysis/gluconeogenesis pathway in Phlebia sp. MG-60, but only 18 in P. chrysosporium.
Orthologous analysis of Phlebia sp. MG-60 and P. chrysosporium
According to the results of orthologous analysis, 5,391 orthologous gene pairs of Phlebia sp. MG-60 and P. chrysosporium were acquired. We obtained 729 (FDR < 0.05) orthologous gene pairs of the two fungi, 1,195 significantly differentially expressed in Phlebia sp. MG-60 (FDR < 0.05) and none with any significant difference in the P. chrysosporium (FDR > 0.05) orthologous gene pairs (Additional file 9: Table S7). Next, we compared the orthologous genes related to glycolysis/gluconeogenesis based on the KEGG pathway database for Phlebia sp. MG-60 and P. chrysosporium. As shown in Fig. 2a, five orthologous genes annotated as glyceraldehyde 3-phosphate dehydrogenase, phosphoglycerate kinase, pyruvate decarboxylase (PDC), phosphoglucomutase, and 2,3-bisphosphoglycerate-independent phosphoglycerate mutase by BLAST search were consistently upregulated, and two orthologous genes annotated aldose 1-epimerase and phosphoenolpyruvate carboxykinase (ATP) were downregulated in both Phlebia sp. MG-60 and P. chrysosporium. Two orthologous genes showed different expression levels in Phlebia sp. MG-60 and P. chrysosporium, and only a pyruvate kinase gene was particularly upregulated in Phlebia sp. MG-60. Twelve orthologous gene pairs, including genes encoding aldehyde dehydrogenase, fructose-1,6-bisphosphatase, pyruvate dehydrogenase E1 component (two genes), glucose-6-phosphate isomerase, pyruvate dehydrogenase E2 component, dihydrolipoamide dehydrogenase, hexokinase (two genes), alcohol dehydrogenase (ADH), fructose-bisphosphate aldolase, and 6-phosphofructokinase, were significantly differentially expressed in Phlebia sp. MG-60 (FDR < 0.05), with no significant difference in P. chrysosporium (FDR > 0.05) (Fig. 2b).

Comparison of Phlebia sp. MG-60 and P. chrysosporium orthologs. The X-axis indicates log fold change of Phlebia sp. MG-60, and the Y-axis indicates log fold change of P. chrysosporium. a: Orthologous genes with significantly different expression (FDR < 0.05) in Phlebia sp. MG-60 and P. chrysosporium. Orthologous genes related to glycolysis/gluconeogenesis based on the KEGG pathway database are shown in red, including 1: glucose-6-phosphate 1-epimerase [EC: 5.1.3.15]; 2: phosphoglucomutase [EC: 5.4.2.2]; 3: pyruvate decarboxylase [EC: 4.1.1.1]; 4: phosphoglycerate kinase [EC: 2.7.2.3]; 5: glyceraldehyde 3-phosphate dehydrogenase [EC: 1.2.1.12]; 6: 2,3-bisphosphoglycerate-independent phosphoglycerate mutase [EC: 5.4.2.12]; 7: aldose 1-epimerase [EC: 5.1.3.3]; 8: phosphoenolpyruvate carboxykinase (ATP) [EC: 4.1.1.49]; 9: pyruvate kinase [EC: 2.7.1.40]. b: Significantly differentially expressed in Phlebia sp. MG-60 (FDR < 0.05) but no significant difference for P. chrysosporium (FDR > 0.05) orthologous genes. Orthologous genes related to glycolysis/gluconeogenesis based on the KEGG pathway database are shown in red, including 1: aldehyde dehydrogenase [EC: 1.2.1.3]; 2: fructose-1,6-bisphosphatase I [EC: 3.1.3.11]; 3: pyruvate dehydrogenase E1 component [EC: 1.2.4.1]; 4: glucose-6-phosphate isomerase [EC: 5.3.1.9]; 5: pyruvate dehydrogenase E1 component [EC: 1.2.4.1]; 6: pyruvate dehydrogenase E2 component [EC: 2.3.1.12]; 7: dihydrolipoamide dehydrogenase [EC: 1.8.1.4]; 8: hexokinase [EC: 2.7.1.1]; 9: alcohol dehydrogenase, propanol-preferring [EC: 1.1.1.1]; 10: hexokinase [EC: 2.7.1.1]; 11: fructose-bisphosphate aldolase [EC: 4.1.2.13]; 12: 6-phosphofructokinase [EC: 2.7.1.11]
Semi-quantitative RT-PCR and qRT-PCR of Phlebia sp. MG-60
To validate the reliability of the expression profiles obtained by RNA-seq, expression levels of five highly expressed genes related to ethanol fermentation in Phlebia sp. MG-60, including one sugar transporter (TR10028|c0_g1), three genes mapped to the glycolysis/gluconeogenesis pathway (TR8916|c1_g1, TR11270|c0_g1, TR9324|c0_g1), and one ADH (TR11797|c0_g1), were compared by semi-quantitative RT-PCR and qRT-PCR. As shown in Fig. 3a, semi-quantitative RT-PCR indicated that all of these genes showed higher expression at 2 days of incubation. Using qRT-PCR, we examined the expression of these genes at 2 and 9 days of incubation and normalized expression to the actin and HMBS genes (Fig. 3b). These results all indicated that expression levels measured by semi-quantitative RT-PCR and qRT-PCR correlated with the RNA-seq analysis.

Confirmation of MiSeq results by qRT-PCR (a) and RT-PCR (b). Five up-regulated genes of Phlebia sp. MG-60 at 2 days of incubation were selected for confirmation. Actin and hydroxymethylbilane synthase genes were used as reference genes for qRT-PCR. Values are the means ± SD of triplicate samples
Discussion
In this study, we analyzed differential gene expression of the ethanol producing white-rot fungus Phlebia sp. MG-60 under fermenting and non-fermenting conditions using next-generation sequencing techniques, and identified the genes involved in its high ethanol production.
Gene expression of ligninolytic enzymes in Phlebia sp. MG-60
Biological delignification is currently attracting much attention as an alternative technology to traditional physicochemical methods for the saccharification of lignocellulosic biomass [3]. White-rot fungi have a unique ability to degrade lignin via extracellular ligninolytic enzymes such as lignin peroxidase, manganese peroxidase (MnP), and laccase [37]. MnP oxidizes Mn2+ to Mn3+, and Mn3+ acts on monomeric phenol, phenolic lignin dimers, and synthetic lignin [38, 39]. Laccases are a group of multi-copper oxidases, which have the ability to oxidize both phenolic and non-phenolic lignin units [40, 41]. Following BLASTx searches of Phlebia sp. MG-60 against amino acid sequences in the Swiss-Prot database, thirteen MnP genes and eleven laccase genes were detected (Additional file 4: Table S3). Recently, MnP gene transformants of Phlebia sp. MG-60 showed higher MnP activity, and overexpression of the MnP gene improved delignification ability of Phlebia sp. MG-60 [42]. The present study was done using glucose as carbon source and therefore not adapted to discuss about ligninolytic enzymes involved in lignocellulose conversion. However, Phlebia sp. MG-60 is a candidate for an integrated fungal fermentation process due to its efficient delignification.
Expression of glucose transporter gene in Phlebia sp. MG-60
GO functional enrichment analysis of Phlebia sp. MG-60 indicated that two groups, “integral component of membrane” (GO: 0016021) and “membrane” (GO: 0016020), were significantly enriched in the cellular components (Table 2). The first step of sugar metabolism is its transport across the cell membrane [43]. Efficient sugar uptake through the expression of hexose transporter genes can improve fermentation of lignocellulosic biomass to ethanol [44]. Yeast hexose transporters have been most extensively researched, and Saccharomyces cerevisiae has at least eight hexose transporters that mediate the uptake of glucose [45, 46]. In the present study, nine glucose transporter genes were expressed in Phlebia sp. MG-60. In particular, TR10228|c0_g1, TR10117|c0_g1, TR2714|c0_g1 were characterized as high-affinity glucose transporters, and their expression increased 47.8-, 12.9-, and 4.9-fold under fermenting conditions (Additional file 4: Table S3). Ali et al. reported that the overexpression of a high-affinity glucose transporter gene in the fungus Fusarium oxysporum directly affected the glucose and xylose transport capacity and ethanol yield [47]. These results confirm that high-affinity glucose transporter genes expressed at a high level in Phlebia sp. MG-60 play an important role in the initiation of ethanol fermentation from glucose.
Expression of genes involved in glycolysis
DEGs involved in the glycolysis pathway and ethanol fermentation in Phlebia sp. MG-60 are summarized in Fig. 4. For all steps in glycolysis, genes were upregulated in Phlebia sp. MG-60 under fermenting conditions, including hexokinase [EC: 2.7.1.1], glucose-6-phosphate isomerase [EC: 5.3.1.9], 6-phosphofructokinase [EC: 2.7.1.11], fructose-bisphosphate aldolase [EC: 4.1.2.13], triosephosphate isomerase [EC: 5.3.1.1], glyceraldehyde 3-phosphate dehydrogenase [EC: 1.2.1.12], phosphoglycerate kinase [EC: 2.7.2.3], 2,3-bisphosphoglycerate-independent phosphoglycerate mutase [EC: 5.4.2.12], enolase [EC: 4.2.1.11], and pyruvate kinase [EC: 2.7.1.40] (Fig. 4, Additional file 7: Table S5). KEGG annotations indicated that all genes involved in the production of ethanol from glucose were expressed in Phlebia sp. MG-60 (Additional file 6: Figure S2). In contrast, only nine of these genes were expressed in P. chrysosporium (Additional file 6: Figure S2). In current commercial ethanol production, the yeast S. cerevisiae is mostly researched, which shows high ethanol yield from glucose, and its prime metabolic pathway of ethanol fermentation is glycolysis [48]. Our results clarified that the ethanol producing white-rot fungus Phlebia sp. MG-60 produces ethanol through the same glycolytic pathway, and that it produces ethanol efficiently by upregulating all genes involved in glycolysis.

Summary of upregulated DEGs of Phlebia sp. MG-60 involved in glycolysis and ethanol fermentation. A list of the identified enzymes is provided in Additional file 7: Table S5
Orthologous analysis between Phlebia sp. MG-60 and P. chrysosporium
To determine whether the expression levels of gene orthologs between Phlebia sp. MG-60 and P. chrysosporium are correlated, we performed orthologous analysis using FastOrtho. From the results, glyceraldehyde 3-phosphate dehydrogenase [EC: 1.2.1.12], phosphoglycerate kinase [EC: 2.7.2.3], PDC [EC: 4.1.1.1], phosphoglucomutase [EC: 5.4.2.2], and 2,3-bisphosphoglycerate-independent phosphoglycerate mutase [EC: 5.4.2.12], which are important in glycolysis, as shown in Fig. 4, were consistently upregulated in both Phlebia sp. MG-60 and P. chrysosporium under fermenting condition (Fig. 2a). In particular, almost all log Fold Change (FC) values of upregulated orthologs in Phlebia sp. MG-60, except for phosphoglucomutase, were much higher than in P. chrysosporium. On the other hand, aldose 1-epimerase [EC: 5.1.3.3] and phosphoenolpyruvate carboxykinase [EC: 4.1.1.49] were downregulated in both species under fermenting condition. Glucose-6-phosphate 1-epimerase [EC: 5.1.3.15] was upregulated in P. chrysosporium but downregulated in Phlebia sp. MG-60 (Fig. 2a). Although these three enzymes mapped to the glycolysis/gluconeogenesis pathway, they do not play a critical role in glycolysis. Only pyruvate kinase [EC: 2.7.1.40] was upregulated in Phlebia sp. MG-60 but downregulated in P. chrysosporium. This observation suggested that pyruvate kinase is a rate-limiting enzyme in ethanol fermentation by white-rot fungi. We also compared significantly differentially expressed genes in Phlebia sp. MG-60 (FDR < 0.05) and genes that showed no significant difference for orthologous gene pairs in P. chrysosporium (FDR > 0.05) (Fig. 2b). Six genes involved in glycolysis and ethanol fermentation were significantly differentially upregulated in Phlebia sp. MG-60, including glucose-6-phosphate isomerase [EC: 5.3.1.9], hexokinase [EC: 2.7.1.1] (two genes), fructose-bisphosphate aldolase [EC: 4.1.2.13], 6-phosphofructokinase [EC: 2.7.1.11], and ADH [EC: 1.1.1.1]. Aldehyde dehydrogenase [EC: 1.2.1.3] was differentially downregulated in Phlebia sp. MG-60, so it may be related to ethanol production.
Expression of genes involved in ethanol fermentation via pyruvate
We identified upregulated genes involved in ethanol fermentation via pyruvate in Phlebia sp. MG-60 (Fig. 4, Additional file 7: Table S5). Two genes coding for PDC [EC: 4.1.1.1] were identified as DEGs catalyzing the conversion of pyruvate into acetaldehyde and carbon dioxide. Finally, ADH [EC: 1.1.1.1] converts acetaldehyde to ethanol. It is common knowledge that there are two ethanol fermentation pathways from pyruvate. In the two-step ethanol fermentation pathway, pyruvate is non-oxidatively decarboxylated to acetaldehyde by PDC, and then acetaldehyde is converted to ethanol by ADH [49]. In the three-step ethanol fermentation pathway, pyruvate is oxidatively decarboxylated to acetyl-CoA by pyruvate ferredoxin oxidoreductase and pyruvate formate lyase. Acetyl-CoA is then converted to acetaldehyde by a CoA-dependent-acetylating acetaldehyde dehydrogenase. Finally, ADH converts acetaldehyde to ethanol [50, 51]. The three-step pathway is widespread in bacteria, but not in white-rot fungi. In the present study, pyruvate ferredoxin oxidoreductase, pyruvate formate lyase, and CoA-dependent-acetylating acetaldehyde dehydrogenase were not detected in the DEGs of Phlebia sp. MG-60 under fermenting conditions. These observations indicated that Phlebia sp. MG-60 mainly uses the two-step pathway of ethanol production from pyruvate, and that the PDC and ADH genes play the major roles in ethanol production. Recently, we reported that highly expressing transformants of the PDC gene in the white-rot fungus P. sordida YK-624 showed improved ethanol production [52]. As shown in Additional file 7: Table S5 and Additional file 8: Table S6, two genes of PDC in Phlebia sp. MG-60 increased PDC expression by 5.0- and 6.3-fold, and three ADH genes increased ADH expression by 11.6-, 4.4-, and 4.1-fold under fermenting conditions. Although six ADH genes were identified in this study, three were downregulated under fermenting conditions. These three upregulated ADH genes may have been the main ones used for ethanol production in the fermentation conditions of this study. In contrast, PDC expression was 2.9- and 3.0-fold increased, and ADH expression was 2.9-fold decreased in P. chrysosporium based on RNA-seq data. Additionally, eleven genes for aldehyde dehydrogenase [EC: 1.2.1.3], which is responsible for the subsequent oxidation of acetaldehyde into acetate, were identified in Phlebia sp. MG-60. However, all aldehyde dehydrogenase genes of Phlebia sp. MG-60 showed low expression in fermentation conditions (Additional file 4: Table S3), which suggested that low expression of aldehyde dehydrogenase in Phlebia sp. MG-60 might be a cause of high ethanol productivity.
During ethanol fermentation, the ethanol produced inhibits the growth and viability of the microorganism [53, 54]. Thus, a high level of ethanol tolerance is considered to be important for a high yield of ethanol. It has been reported that alteration in ethanol tolerance could be affected by many factors, such as fatty acid composition and activity of plasma membrane H+-ATPase [55–58]. In this study, “fatty acid biosynthetic process” (GO: 0006633) in the biological processes category and “ATP binding” (GO: 0005524) in the molecular functions were the most abundant terms in GO enrichment analysis of Phlebia sp. MG-60 under fermenting conditions (Table 2). These observations indicated that a high resistance of Phlebia sp. MG-60 to ethanol stress might have an effect on its high ethanol productivity.
Conclusions
In this study, we provided novel transcriptomic information on the ethanol producing white-rot fungus Phlebia sp. MG-60, and investigated the genes involved in its high ethanol yield by comparing them to the model white-rot fungus P. chrysosporium. Based on differential gene expression analysis, the genes involved in glucose transport, glycolysis, and ethanol fermentation were upregulated in Phlebia sp. MG-60. This suggests that the high ethanol productivity of Phlebia sp. MG-60 is due to genes related to glucose uptake, metabolism, including the production of pyruvate, and ethanol synthesis, which are upregulated under fermenting conditions. This study may lead to a better understanding of fermentation by white-rot fungi, and will provide information needed for the genetic engineering of ethanol production.
Abbreviations
ADH, alcohol dehydrogenase; ATP, phosphoenolpyruvate carboxykinase; DEGs, Differentially expressed genes; FDR, False Discovery Rate; GO, Gene ontology; HMBS: hydroxymethylbilane synthase; HPLC, high-performance liquid chromatography; KEGG, Kyoto Encyclopedia of Genes and Genomes; MnP, manganese peroxidase; ORFs, Open reading frames; PDA, potato dextrose agar; PDC, pyruvate decarboxylase; qRT-PCR, quantitative real-time reverse transcription PCR; RNA-seq, RNA-sequencing
References
Wang GS, Pan XJ, Zhu JY, Gleisner R, Rockwood D. Sulfite pretreatment to overcome recalcitrance of lignocellulose (SPORL) for robust enzymatic saccharification of hardwoods. Biotechnol Prog. 2009;25:1086–93.
Hatakka A, Hammel KE: Fungal biodegradation of lignocelluloses. In: Industrial Applications. Edited by Hofrichter M, vol. 10. Berlin, Heidelberg: Springer Berlin Heidelberg; 319-340. http://link.springer.com/chapter/10.1007%2F978-3-642-11458-8_15.
Moreno AD, Ibarra D, Alvira P, Tomás-Pejó E, Ballesteros M. A review of biological delignification and detoxification methods for lignocellulosic bioethanol production. Crit Rev Biotechnol. 2015;35:342–54.
Dashtban M, Schraft H, Qin W. Fungal bioconversion of lignocellulosic residues; opportunities & perspectives. Int J Biol Sci. 2010;5:578–95.
Wan C, Li Y. Effectiveness of microbial pretreatment by Ceriporiopsis subvermispora on different biomass feedstocks. Bioresour Technol. 2011;102:7507–12.
le Khuong D, Kondo R, De Leon R, Anh TK, Meguro S, Shimizu K, Kamei I. Effect of chemical factors on integrated fungal fermentation of sugarcane bagasse for ethanol production by a white-rot fungus, Phlebia sp. MG-60. Bioresour Technol. 2014;167:33–40.
Lundell TK, Mäkelä MR, de Vries RP, Hildén KS. Genomics, lifestyles and future prospects of wood–decay and litter–decomposing basidiomycota. Adv Bot Res. 2014;70:329–70.
Binder M, Justo A, Riley R, Salamov A, Lopez-Giraldez F, Sjokvist E, Copeland A, Foster B, Sun H, Larsson E, Larsson KH, Townsend J, Grigoriev IV, Hibbett DS. Phylogenetic and phylogenomic overview of the Polyporales. Mycologia. 2013;105:1350–73.
Ruiz-Dueñas FJ, Lundell T, Floudas D, Nagy LG, Barrasa JM, Hibbett DS, Martínez AT. Lignin-degrading peroxidases in Polyporales: an evolutionary survey based on 10 sequenced genomes. Mycologia. 2013;105:1428–44.
Rytioja J, Hildén K, Yuzon J, Hatakka A, de Vries RP, Mäkelä MR. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol Mol Biol Rev. 2014;78:614–49.
Okamoto K, Imashiro K, Akizawa Y, Onimura A, Yoneda M, Nitta Y, Maekawa N, Yanase H. Production of ethanol by the white-rot basidiomycetes Peniophora cinerea and Trametes suaveolens. Biotech Lett. 2010;32:909–13.
Okamoto K, Nitta Y, Maekawa N, Yanase H. Direct ethanol production from starch, wheat bran and rice straw by the white rot fungus Trametes hirsute. Enzyme Microb Technol. 2011;48:273–7.
Okamoto K, Uchii A, Kanawaku R, Yanase H. Bioconversion of xylose, hexoses and biomass to ethanol by a new isolate of the white rot basidiomycete Trametes versicolor. Springerplus. 2014;3:121.
Li X, Kondo R, Sakai K. Studies on hypersaline-tolerant white-rot fungi I: screening of lignin-degrading fungi in hypersaline conditions. J Wood Sci. 2002;48:147–52.
Kamei I, Hirota Y, Meguro S. Integrated delignification and simultaneous saccharification and fermentation of hard wood by a white-rot fungus, Phlebia sp. MG-60. Bioresour Technol. 2012;126:137–41.
Kamei I, Hirota Y, Mori T, Hirai H, Meguro S, Kondo R. Direct ethanol production from cellulosic materials by the hypersaline-tolerant white-rot fungus Phlebia sp. MG-60. Bioresour Technol. 2012;112:137–42.
Garg R, Sahoo A, Tyagi AK, Jain M. Validation of internal control genes for quantitative gene expression studies in chickpea. Biochem Biophy Res Commun. 2010;396:283–8.
Martin M. Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet J. 2011;1(17):10–2.
Pearson WR, Wood T, Zhang Z, Miller W. Comparison of DNA sequences with protein sequences. Genomics. 1997;46:24–36.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–512.
Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range ofexpression levels. Bioinformatics. 2012;28:1086–92.
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, Griffith M, Raymond A, Thiessen N, Cezard T, Butterfield YS, Newsome R, Chan SK, She R, Varhol R, Kamoh B, Prabhu A-L, Tam A, Zhao Y, Moore RA, Hirst M, Marra MA, Jones SJM, Hoodless PA, Birol I. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7:909–12.
Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, Zhou X, Lam T-W, Li Y, Xu X, Wong GK, Wang J. SOAPdenovo-Trans de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014;30:1660–6. doi:10.1093/bioinformatics/btu077.
Li B, Fillmore N, Bai Y, Collins M, Thomson JA, Stewart R, Dewey CN. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2014;15:553.
Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–14.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Li B, Dewey C. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Bairoch A, Boeckmann B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 1991;19:2247–9.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14.
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–61.
Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP, Dalay O, Driscoll T, Hix D, Mane SP, Mao C, Nordberg EK, Scott M, Schulman JR, Snyder EE, Sullivan DE, Wang C, Warren A, Williams KP, Xue T, Yoo HS, Zhang C, Zhang Y, Will R, Kenyon RW, Sobral BW. Patric: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun. 2011;79:4286–98.
Dohra H, Tanaka K, Suzuki T, Fujishima M, Suzuki H. Draft genome sequences of three Holospora species (Holospora obtusa, Holospora undulata, and Holospora elegans), endonuclear symbiotic bacteria of the ciliate Paramecium caudatum. FEMS Microbiol Lett. 2014;359:16–8.
Li L, Stoeckert Jr CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Yamada Y, Wang J, Kawagishi H, Hirai H. Improvement of ligninolytic properties by recombinant expression of glyoxal oxidase gene in hyper lignin-degrading fungus Phanerochaete sordida YK-624. Biosci Biotechnol Biochem. 2014;78:2128–33.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–96.
Lundell TK, Mäkelä MR, Hildén K. Lignin-modifying enzymes in filamentous basidiomycetes--ecological, functional and phylogenetic review. J Basic Microbiol. 2010;50:5–20.
Gold MH, Alic M. Molecular biology of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Microbiol Rev. 1993;57:605–22.
Martínez AT, Speranza M, Ruiz-Dueñas FJ, Ferreira P, Camarero S, Guillén F, Martínez MJ, Gutiérrez A, del Río JC. Biodegradation of lignocellulosics: microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int Microbiol. 2005;8:195–204.
Bourbonnais R, Paice MG. Oxidation of non-phenolic substrates. An expanded role for laccase in lignin biodegradation. FEBS Lett. 1990;267:99–102.
Mayer AM, Staples RC. Laccase: new functions for an old enzyme. Phytochemistry. 2002;60:551–65.
Yamasaki Y, Yamaguchi M, Yamagishi K, Hirai H, Kondo R, Kamei I, Meguro S. Expression of a manganese peroxidase isozyme 2 transgene in the ethanologenic white rot fungus Phlebia sp. strain MG-60. Springerplus. 2014;3:699.
Ozcan S, Johnston M. Function and regulation of yeast hexose transporters. Microbiol Mol Biol Rev. 1999;63:554–69.
Van Vleet JH, Jeffries TW. Yeast metabolic engineering for hemicellulosic ethanol production. Curr Opin Biotechnol. 2009;20:300–6.
Sedlak M, Ho NW. Characterization of the effectiveness of hexose transporters for transporting xylose during glucose and xylose co-fermentation by a recombinant Saccharomyces yeast. Yeast. 2004;21:671–84.
Young EM, Comer AD, Huang H, Alper HS. A molecular transporter engineering approach to improving xylose catabolism in Saccharomyces cerevisiae. Metab Eng. 2012;14:401–11.
Ali SS, Nugent B, Mullins E, Doohan FM. Insights from the fungus Fusarium oxysporum point to high affinity glucose transporters as targets for enhancing ethanol production from lignocellulose. PLoS One. 2013;8:e54701.
Bai FW, Anderson WA, Moo-Young M. Ethanol fermentation technologies from sugar and starch feedstocks. Biotechnol Adv. 2008;26:89–105.
Ohta K, Beall DS, Mejia JP, Shanmugam KT, Ingram LO. Genetic improvement of Escherichia coli for ethanol production: chromosomal integration of Zymomonas mobilis genes encoding pyruvate decarboxylase and alcohol dehydrogenase II. Appl Environ Microbiol. 1991;57:893–900.
Burdett D, Zeikus JG. Purification of acetaldehyde dehydrogenase and alcohol dehydrogenases from Thermoanaerobacter ethanolicus 39E and characterization of the secondary-alcohol dehydrogenase (2 degrees Adh) as a bifunctional alcohol dehydrogenase--acetyl-CoA reductive thioesterase. Biochem J. 1994;302:163–70.
Yao S, Mikkelsen MJ. Identification and overexpression of a bifunctional aldehyde/alcohol dehydrogenase responsible for ethanol production in Thermoanaerobacter mathranii. J Mol Microbiol Biotechnol. 2010;19:123–33.
Wang J, Hirabayashi S, Mori T, Kawagishi H, Hirai H. Improvement of ethanol production by recombinant expression of pyruvate decarboxylase in the white-rot fungus Phanerochaete sordida YK-624. J Biosci Bioeng. 2016;122(1):17–21.
Piper PW. The heat shock and ethanol stress responses of yeast exhibit extensive similarity and functional overlap. FEMS Microbiol Lett. 1995;134:121–7.
Bai FW, Chen LJ, Zhang Z, Anderson WA, Moo-Young M. Continuous ethanol production and evaluation of yeast cell lysis and viability loss under very high gravity medium conditions. J Biotechnol. 2004;110:287–93.
Aguilera F, Peinado RA, Millán C, Ortega JM, Mauricio JC. Relationship between ethanol tolerance, H+ -ATPase activity and the lipid composition of the plasma membrane in different wine yeast strains. Int J Food Microbiol. 2006;110:34–42.
Supply P, de Kerchove d'Exaerde A, Roganti T, Goffeau A, Foury F. In-frame recombination between the yeast H(+)-ATPase isogenes PMA1 and PMA2: insights into the mechanism of recombination initiated by a double-strand break. Mol Cell Biol. 1995;15:5389–95.
Koukou AI, Tsoukatos D, Drainas C. Effect of ethanol on the phospholipid and fatty acid content of Schizosaccharomyces pombe membranes. J Gen Microbiol. 1990;136:1271–7.
Henderson CM, Block DE. Examining the role of membrane lipid composition in determining the ethanol tolerance of Saccharomyces cerevisiae. Appl Environ Microbiol. 2014;80:2966–72.
Availability of supporting data
The data sets supporting the results of this article are available in the DDBJ Sequence Read Archive (DRA) (accession numbers for the sequences of Phlebia sp. MG-60 and P. chrysosporium are DRA004667 and DRA004668, respectively).
Authors’ contributions
HH conceived the project and designed outlines most of the experiments. JW, TS, HD and ST performed RNA-Seq and analysis of the resulting data. IK provided Phlebia sp. MG-60. HD, TM and HKaw gave advice on the experiments, data analysis. JW performed other experiments and analyzed data with HKak’s and SA’s assistance. JW, TS and HH wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
This manuscript has no financial or non-financial competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Primers used for qRT-PCR and semi-quantitative RT-PCR. (DOCX 29 kb)
Additional file 2: Figure S1.
Time courses of mycelial growth and pH changes in liquid cultures of Phlebia sp. MG-60 (A) and P. chrysosporium (B). (PPTX 153 kb)
Additional file 3: Table S2.
Number of sequencing reads. (DOCX 23 kb)
Additional file 4: Table S3.
Data for Phlebia sp. MG-60 transcripts. (XLSX 3497 kb)
Additional file 5: Table S4.
Data for P. chrysosporium transcripts. (XLSX 2407 kb)
Additional file 6: Figure S2.
Transcripts of Phlebia sp. MG-60 and of P. chrysosporium mapped to the glycolysis/gluconeogenesis pathway based on the KEGG pathway database. A list of the identified enzymes is provided in Tables S5 and S6. Genes showing up-regulation and down-regulation are boxed in red and green, respectively. The number of genes is shown in blue. (DOCX 161 kb)
Additional file 7: Table S5.
Fold changes of transcripts mapped to the glycolysis/gluconeogenesis pathway of Phlebia sp. MG-60 based on KEGG. (DOCX 35 kb)
Additional file 8: Table S6.
Fold changes of transcripts mapped to the glycolysis/gluconeogenesis pathway of P. chrysosporium based on KEGG. (DOCX 31 kb)
Additional file 9: Table S7.
Orthologous analysis of Phlebia sp. MG-60 and P. chrysosporium. (XLSX 6512 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, J., Suzuki, T., Dohra, H. et al. Analysis of ethanol fermentation mechanism of ethanol producing white-rot fungus Phlebia sp. MG-60 by RNA-seq. BMC Genomics 17, 616 (2016). https://doi.org/10.1186/s12864-016-2977-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-2977-7