
Abstract
Background
Pseudostellaria heterophylla produces both closed (cleistogamous, CL) and open (chasmogamous, CH) flowers on the same individual but in different seasons. The production of CH and CL flowers might be in response to environmental changes. To better understand the molecular mechanisms of CH and CL flowering, we compared the transcriptome of the two types of flowers to examine differential gene expression patterns, and to identify gene regulatory networks that control CH and CL flowering.
Results
Using RNA sequencing, we identified homologues of 428 Arabidopsis genes involved in regulating flowering processes and estimated the differential gene expression patterns between CH and CL flowers. Some of these genes involved in gene regulatory networks of flowering processes showed significantly differential expression patterns between CH and CL flowers. In addition, we identified another 396 differentially expressed transcripts between CH and CL flowers. Some are involved in environmental stress responses and flavonoid biosynthesis.
Conclusions
We propose how the differential expression of key members of three gene regulatory modules may explain CH and CL flowering. Future research is needed to investigate how the environment impinges on these flowering pathways to regulate CH and CL flowering in P. heterophylla.
Similar content being viewed by others
Background
Cleistogamy is a special mating system of flowering plants and has been reported for approximately 700 species [1–3]. Three categories of cleistogamy are recognized: dimorphic cleistogamy, complete cleistogamy and induced cleistogamy [3]. Dimorphic cleistogamous plants produce both closed (cleistogamous, CL) flowers and open (chasmogamous, CH) flowers on the same individual or on different individuals. The CL flower is often more juvenile in appearance and is characterized mainly by reductions in flower size, corolla and stamen number. A CL flower is also considered to be a modified form of a CH flower [1, 4–7]. Complete cleistogamous plants produce only CL flowers, whereas induced cleistogamous plants can produce both CL and CH flowers but without any floral organ reduction in the CL flowers. The CL flowers are induced due to the failure of the flowers to open when changes in certain environmental factors [3]. In several cleistogamous species, the ability to produce CH and CL flowers can be affected by seasonal and environmental stresses, including drought, flood, salinity, nutrient deprivation, and shade [1, 3, 8–10]. Cleistogamy may be advantageous because CL flowers ensure seed set by self-pollination under severe environmental conditions [1, 11]. The energetic costs (involved in producing sepals, petals and nectar) of CL flowers appear to be considerably lower than those of CH flowers [12, 13].
The genetic regulation and mechanisms of cleistogamy have been studied in several crops that are not naturally cleistogamous. Cleistogamous flowers in barley and rice lack lodicules or display lodicule deformities due to mutations in certain floral identity genes. A mutation in the ERECT PANICLE2 gene is responsible for the rice cleistogamy mutant cl7(t), in which the lodicules exhibit a weak swelling ability, resulting in the failure of flowers to open [14]. The barley Cleistogamy1 (Cly1) gene is a homolog of A-class APETALA2 (AP2) and is targeted by microRNA miR172. The down-regulation of barley Cly1 caused by a mutation in the miR172 targeting site represses the development of lodicules, thereby leading to cleistogamy [15]. The Bn-CLG1A (CLG for cleistogamy) gene encoding a RINGv E3 ubiquitin ligase was isolated from a Brassica napusmutant; this gene leads to the formation of petal-closed flowers [16].
However, few studies investigated the genetic regulation of CH and CL flowering patterning in naturally cleistogamous species. The genetic regulatory mechanisms of natural cleistogamy may be quite different from those in crops using rare mutants. In naturally cleistogamous plants, both CH and CL flowers are produced by the same individual exposed to different environments [1–3, 17, 18]. Although the appearance of morphologically reduced CL flowers is not caused by the mutation of certain genes, it may be caused by variations in gene expression in some gene regulatory networks (GRNs) controlling the flowering processes. In a cross-species microarray analysis, Morinage et al. [18] identified 69 genes, including those related to floral development and cold stress response that were differentially expressed in CH and CL flowers of an annual cleistogamous herb, Cardamine kokaiensisYahara.
Pseudostellaria heterophylla (Miq.) Pax (Caryophyllaceae) is a native perennial herb widely distributed in north-eastern and eastern China [19, 20], and its roots are used as one of the most popular traditional Chinese medicines. P. heterophylla demonstrates typical dimorphic cleistogamy, usually producing both CH and CL flowers on the same individual plant that appear in different positions and indifferent seasons (CH in spring and CL in summer). This species provides an excellent model for investigating the gene regulatory networks that control CH and CL flowering.
In this study, the RNA-Seq platform based on Illumina Hiseq technology was used to compare the transcriptome of P. heterophylla CH and CL flowers to investigate differences in gene transcription patterns relating to CH and CL flowering processes. The aims of this study were (1) to identify differentially expressed genes involved in the transition from CH to CL flowering in this dimorphic cleistogamous species, and (2) to investigate putative gene regulatory pathways that might determine differences between CH and CL flowering patterning and processes. Because cleistogamous plants produce CL flowers adapted to severe environmental conditions, our study may help to shed light on the molecular basis for the evolution of environmentally-dependent plant mating systems.
Results
Morphological characters of CH and CL flower in P. heterophylla
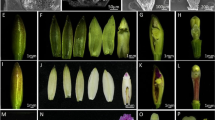
CH flowers of P. heterophylla appear in early spring (late March to late April), when the deciduous trees and shrubs begin to grow new leaves (Fig. 1a). Mature CH flowers develop at the apical shoot and are composed of four whorls of floral organs: five green sepals, five white petals, 10 stamens, and a single ovary with three stigmas (Fig. 1b, c). CL flowers of P. heterophylla appear in late spring to summer (late April to late June), after the withering of CH flowers and once light levels fall as the forest canopy forms (Fig. 1e). Mature CL flowers develop at the axils of the first several nodes of the shoot, are smaller than CH flowers and are composed of three whorls of floral organs (usually without a petal whorl) that include four purple-red sepals, two stamens, and a single ovary with two stigmas (Fig. 1f, g). The initiation of floral organ primordia is also different between CH and CL flowers. In CH flowers, 5 sepal primordia, 10 stamen primordia, 5 petal primordiaand 3 carpel primordia are initiated (Fig. 1d), whereas 4 sepal primordia, 2 stamen primordia and 2 carpel primordia are initiated in CL flowers (Fig. 1h) [7].

Two types of flower of Pseudostellaria heterophylla. a-d Chasmogamous (CH) flowering. a Habitat of P. heterophylla when producing CH flower. b P. heterophylla individuals with CH flowers. c CH flower is at the apical shoot. d SEM images of the early stage of CH flower development, with initiation of floral organs (S = sepal, A = stamen, G = carpel, 1–10 indicate the identified number of floral organs). e–h Cleistogamous (CL) flowering. e Habitat of P. heterophylla when producing CL flower. f P. heterophylla individuals with CL flowers (CL flowers are under the leaves and cannot be seen). g CL flower is in a lateral position on the stem (black arrows). h SEM images of the early stage of CL flower development, with initiation of floral organs. Bars show 5 mm in c and g, Bars show 40 μm in d and h
Sequencing and assembly
High-throughput sequencing of normalized cDNA libraries resulted in 4,459 million clean pair-end reads for P. heterophylla CH flowers and 4,550 million for CL flowers. The raw sequence files have been uploaded to the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra/) under the study accession number SRP052597.
Using high-throughput RNA-seq analysis, we identified 44,603 unigenes in the total transcriptome of P. heterophylla, with an average length of 860 bp (Table 1). The length distributions of the contigs, transcripts and unigenes are provided in Additional file 1. After strict quality control, the total clean pair-end reads of the two libraries were utilized to reveal transcriptomic information for P. heterophylla. Using Trinity, the assembly of clean reads resulted in 218,061 contigs, with an average length of 354 bp, and 27,783 transcripts, with average length of 610 bp. The species that provided most of the top BLAST hits was Vitis vinifera, with 9,496 hits, followed by Populus trichocarpa and Ricinus communis, with 4,724 and 4,269 hits, respectively.
Functional annotation
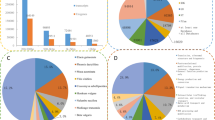
To obtain functional annotations, all of unigenes were BLASTed against the NCBI non-redundant database, and 32,488 unigenes were searched using BLAST in the Swiss-Prot database (Additional file 2). The functional annotations of P. heterophylla genes by the GO, eggNOG and KEGG databases are also shown in Additional file 2. Using the eggNOG database, total unigenes were annotated and subdivided into 25 clusters of orthologous groups (Fig. 2). Among them, 19.93 % (9,854) and 17.90 % (8,850), respectively, of the unigenes obtained from P. heterophylla were assigned to “Function unknown” and “General function prediction only”. These categories were followed by a cluster of “signal transduction mechanisms” (4,014; 8.01 %), “Posttranslational modification, protein turnover, chaperones” (3,621; 7.32 %), and “Transcription” (2,922; 5.91 %); the cluster of “cell mobility” (36; 0.07 %) was the smallest group.

Evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) analysis. A total of 44,603unigenes were analyzed using eggnog database and subdivided into 25 clusters of orthologous (COG) classifications. x-axis, COG classifications; y-axis, number of unigenes
Floral genes are identified
Because one of our aims was to identify candidate genes that are responsible for flowering processes, we specifically searched for P. heterophylla homologues of genes and gene families that are known to be involved in flowering time, floral identity, and flower development in Arabidopsis thaliana. Candidate homologues were identified as BLAST hits. For the Arabidopsis flowering related genes with more than one blast hit to our transcriptome, we chose the longest transcript with the highest identity with Arabidopsis. We identified homologues of 428 Arabidopsis genes involved in regulating flowering processes, sharing 64–83 % nucleic acid identity with the Arabidopsis genes. These candidate flowering-related genes included those affecting flowering time regulation, floral meristem identity, floral organ identity, flower organ development, and establishment of organ and whorl boundaries. Among them were putative homologous genes controlling floral organ identity (ABCE model genes) and some other MADS-box genes (AGAMOUS-like), genes controlling flowering time (FLAVIN-BINDING, KELCH REPEAT, F BOX 1 (FKF1), SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL)), genes involved in meristem size determination and maintenance (CLAVATA2, WUSCHEL and SHOOT MERISTEMLESS), genes involved in organ boundary establishment (CUP-SHAPED COTYLEDON 1), and genes involved in controlling organ number, shape, size, and proportions (PINOID, PETAL LOSS, ROXY1)(Additional file 3, the gene names herein are those used in Arabidopsis) [21–28].
Validation of expression of floral genes involved in flowering gene regulatory networks
The expression of the 24 key candidate genes (Additional file 4) involved in three flowering regulatory modules was validated using qRT-PCR (Fig. 3). The expression patterns of most of loci were consistent with transcriptome sequencing based on the Pearson correlation coefficient value (r = 0.838). FKF1, FT, STM, AP1, AP3, and PI, which were expressed differentially between CH and CL flowers, were detected both by RNA-seq and qRT-PCR (Fig. 3). The remaining flowering genes such as CRY2, PHY, CO, SOC1, AGL24, etc. were not expressed differentially between CH and CL flowers. The expression of GI and CDF were not considered differentially expressed between CH and CL flowers as they were not consistent between RNA-sequencing and qRT-PCR. The expression variation of these genes may play important roles in determining CH and CL flowering, however, as we discuss below.

Comparisons of RNA sequencing and qPCR expression data for twenty-four genes associated with flowering process between CH and CL flowers. The Log fold-change (y-axis) in RNA sequencing was estimated based on the data obtained by Reads per kilobase of exon per million mapped reads values (yellow bars). The log fold-change in qPCR were calculated using the comparative CT method (blue bars). We calculated the Pearson correlation coefficient (r)between the different methods for all transcripts. The correlation coefficient was 0.838, p <0.00001
Differentially expressed transcripts abundance
Comparing the RNA-seq data between CH and CL flowers, we found a total of 396 genes were differentially expressed using the strict cutoff thresholds (fold-change >4, FDR <0.05); Of this total, 93 were up-regulated in CH flowers, 303 were up-regulated in CL flowers, three transcripts were expressed only in CH flowers, and 57 were expressed only in CL flowers (Additional file 5).
The GO enrichment analyses showed that the differentially expressed transcripts (DETs) were enriched in biological process, cellular components and molecular functions (Fig. 4 and Additional file 6) using cutoffs of FDR <0.05. A total of thirteen enriched GO terms were identified, including eight of biological process, three of cellular components and two of molecular functions (Fig. 4). Further, the biological process are related to the response to biotic and abiotic stimulus, the response to endogenous stimulus, the response to external stimulus, the response to stress, and sequence-specific DNA binding transcription activity were attributing to the significant enrichment (p-value <10−3). Moreover, cellular component associated with external encapsulating structure and extracellular region, and molecular function associated with molecular function and binding, were attributing to the enrichment (Fig. 4).

Distributions of differentially expressed transcripts (DETs) (x-axis) into Gene Ontology (GO) categories (y-axis) according to GO enrichment analysis. Up-regulated transcript numbers were shown in bars, red bars representing up-regulated in CH flowers and blue bars representing up-regulated in CL flowers. A p-value and a false discovery rate (FDR) were given for significance
To identify significantly enriched KEGG categories for the DETs from the CH and CL libraries, we assigned 171 DETs to the KEGG database. The details of the KEGG classification are presented in Additional file 7 and 8. The major pathways identified were “metabolism”, “biosynthesis of secondary metabolites”, “hormone signal transduction”, and “plant-pathogen interaction”.
Discussion
This is the first work to analyse the flowering GRNs with RNA-seq based transcriptomes in a naturally cleistogamous plant to understand the genetic mechanisms for different flower patterning. Gene expression controlling development can be modified as a consequence of environmental changes, leading to altered multiple flower developmental pathways. Three modules are proposed to determine CH and CL flower process in natural populations (Fig. 5).

The putative gene regulatory networks (GRNs) that regulate the production of chagamous (CH) and cleistogamous (CL) flowering in Pseudostellaria heterophylla. Genes represented by blocks. Lines with an arrow represent promotion, and those with a perpendicular bar represent repression. a GRNs in CH flower production. b GRNs in CL flower production. Gene expression variation represented by different colour in CL flowering GRNs: gene expression level not to change is shown in white; gene expression level up-regulated is shown in red; gene expression level down-regulated is shown in green. FKF1down-regulated due to the change of Circadian Clock, FT down-regulated due to the poor light quality, the floral identity gene AP1up-regulated due to STM promotion. AP3 and PI down-regulated due to down-regulated upstream genes
The CO-FT module regulates CH and CL flowering time
Day length and the quality of light play a significant role in the floral transition in many plants [21, 22, 25, 28]. The CO-FT module was proposed to explain how plants might initiate flowering in response to photoperiod [21, 22, 28, 29], and this module is highly conserved across plant species [22]. In Arabidopsis, the expression of FT is regulated by the CO protein and is a crucial aspect of photoperiodic flowering. CO expression is associated with the combined action of photoreceptors (PHYA, PHYB, and CRY2) and the circadian clock system (FKF1 and GI) [30–33]. Further, the protein FT, which acts as a universal florigen [34–38], is a major target of CO. Transport of the transcription factor FT from the leaves to the shoot apical meristem results in the activation of transcriptional cascades that specify the transition from vegetative to reproductive growth.
Homologues of most genes associated with the CO-FT module were identified in the P. heterophylla transcriptome, including FKF1, GI, CO, FT, and other factors. Interestingly, the upstream geneFKF1 and the downstream gene FT of the CO-FT module both showed lower expression levels in CL flowers than in CH flowers (Fig. 5). FKF1 has been reported to play a key role in regulating the circadian clock and photoperiodic flowering [30–33]. Searle et al. [39] reported that reducing expression of FT indirectly repressed expression of genes involved in floral induction at the meristem and delayed flowering time in Arabidopsis. The lower levels of FKF1 and FT expression might play important roles in the delay of flowering time in CL flowers in our species. Natural P. heterophylla populations usually grow under temperate forests. CH flowers are produced only in early spring when the deciduous trees and shrubs begin to grow new leaves; the appearance of CL flowers appears a few weeks later, and light levels fall as the forest canopy forms. The photoperiod and circadian clock changes from those in spring might regulate FKF1 expression and further regulate FT gene expression in CL flowers.
The STM-AP1 module regulates CL flower meristem specification
The specification of FM is crucial for successful flower development [40–43]. Two FM identity genes, AP1 and LFY, are conserved in their FM specification function [42, 44–46], and are regulated by multiple factors (Fig. 5).
Homologues of these FM identity genes and their regulatory factors were also identified in the P. heterophylla transcriptome. Surprisingly, although FT and SOC1 expression was reduced in CL flowers, their targetsAP1 and LFY were up-regulated. AP1 is highly expressed in CL flowers, which makes it a good candidate for contributing to the spatial specification of FM initiation. We found that STM, which directly activateAP1 and LFY, was significantly up-regulated in CL flowers (Fig. 5). In Arabidopsis, STM acts to regulate flower patterning, branching, and internode growth and the specification of axillary meristems during inflorescence development [47–51]. In P. heterophylla, CL inflorescences develop in the axils of the first several nodes of the shoot. Unlike CH flower production at the apical meristem, the axillary flower meristems may play a crucial role in the establishment of a CL inflorescence, ultimately leading to the formation of CL flowers. We speculate that STM plays a fundamental role in promoting the formation of CL FMs in the axils of cauline leaves in P. heterophylla.
The ABCE model regulates CH and CL floral patterning
The classic ABC (or ABCE) model proposed to determine floral organ identity is conserved across angiosperms [25–27]. Four classes of genes (A, B, C, and E) are well known to act in combination for proper organ formation in each whorl [52]. All of the floral organ identity genes homologous to those of Arabidopsis A, B, C, and E genes were also identified in P. heterophylla (Fig. 5). The CH flower of P. heterophylla is a typical eudicot pentamerous flower with four whorls: five sepals, five petals, ten stamens and three carpels. However, the P. heterophylla CL flower displays floral organ reductions: sepals are reduced to four; petals are absent; stamens are reduced to two; and carpels are reduced to two (Fig. 1d, h) [7]. The A class gene AP1 is highly expressed in CL flowers. AP1 has two functions, floral meristem specification and perianth identity, and these two functions may not be separable [41, 53, 54]. Our previous study found the significant difference in the initiation sequence of sepals in CH vs CL flowers of P. heterophylla: five sepal primordia are initiated one by one in a 2/5 helix sequence in the CH flower; in contrast, four sepal primordia in the CL flower initiate alternatively in pairs [7]. The four sepals of the CL flower are not derived from sepal reduction of the pentamerous CH flower but may arise as a continuation of bracteole initiation [7]. Increased levels of AP1 expression may strengthen the floral meristem specification function of AP1, resulting in bracteole-like sepal formation in the CL flower.
The B class genes AP3 and PI are significantly decreased in CL flowers. AP3 and PI are closely related MADS paralogous lineages, and their functions in establishing the identity of petals and stamens in floral development are conserved in both eudicots and monocots [55, 56]. In our previous floral organogenesis study [7], we found that, in floral organ identity stages, fewer primordia of stamen and absence of petal primordia in CL flowers (Fig. 1d, h), which might explain the stamen number reduction and absence of petals compared to CH flowers. Mondragon-Palomino et al. [57] found that the complex perianth in orchids is patterned by differential expression of multiple B-class gene paralogs. Zhang et al. [58] suggested that disruption of the petal identity gene AP3-3 is highly correlated with loss of petals in the buttercup family (Ranunculaceae). Thus, the reduced stamen number and absence of petals in CL flowers in our species are likely related to the decreased levels of expression in B class genes (Fig. 5b). However, further investigations for ABCE functions in CL and CH flower patterning are needed.
Other differentially expressed genes in CH and CL flowering
Except for those flowering related genes, 369 transcripts were considered significantly differentially expressed between CH and CL flowers. These differentially expressed genes include those related to environmental stress responses, plant defence response, plant hormone signal transduction, plant development and growth and secondary metabolite biosynthesis. Especially, genes involved in stress response were significantly overrepresented (p-value = 2.01E-21, Fig. 4), with14 and 87 stress-response genes up-regulated in CH and CL flowers, respectively. We could not rule out the possibility that the expression differences of stress-related genes could be a result of the specific experimental design. Nevertheless, sixteen transcripts with homology to the HSP family such as HSP70, HSP83, HSP90, and some small Heat Shock Protein (sHSP) families were detected in CL flowers (Additional file 5, marked with red). HSP induction may reflect differential physiological responses to environmental stresses, such as drought, cold, and high temperature [59–61]. In a previous study, two HSP genes were reported to be up-regulated with cold treatment in Cardamine kokaiensis CL flowers [18]. Further, eight homologues of WRKY transcription factor genes (Additional file 5, marked with purple), which are important regulators of active biotic and abiotic stress responses [62], were significantly up-regulated in CL flowers. It is also evident that some members of the WRKY family may play important regulatory roles in promoting flowering [63–65].
With the KEGG analysis, we found nine genes related to flavonoid biosynthesis pathway were significantly up-regulated in CL flowers (Additional file 5, marked with orange). These included encoding enzymes involved in flavonoid and anthocyanin syntheses, such as Anthocyanidin Synthase, Flavonoid 3′-hydroxylase. Flavonoids are important secondary compounds and the main pigments in flowers [66, 67]. The blue and purple flower colours are derived from flavonoids and anthocyanins [68–70]. The up-regulation of genes related to flavonoid and anthocyanin biosyntheses in CL flowers may result in the purple colour of the sepals, instead of the white colour of the petals in CH flowers.
Conclusions
This is the first work to investigate the flowering GRNs in a naturally cleistogamous plant. Gene expression patterns are modified as a consequence of environmental changes, leading to altered multiple flower developmental pathways. We proposed three modules that regulate flowering time, meristem development and patterning in a natural populations of P. heterophylla. Even though investigations of the spatiotemporal expression patterns of these genes are required to assess their functions in determining CH and CL flowering, our study provided a foundation for dissecting the molecular basis of flowering time and patterns in naturally cleistogamous plants. Cleistogamous plants can be a very important model for studying the adaption and evolution of flowering behaviour.
Methods
Plant materials
Materials for the transcriptome sequencing study were collected from a natural population from Mount Lao, located near the East China Sea on the south-eastern coastline of the Shandong Peninsula in China (120°36′10″E, 36°06′27″N). CH flower buds were collected on 8 April, 2014 (average minimum temperature is 8 °C, average maximum temperature is 15 °C and average precipitation is 36 mm in April; http://qingdao.tianqi.com/laoshan/). CL flower buds were collected on 26 May, 2014 (average minimum temperature is 13 °C, average maximum temperature is 20 °C and average precipitation is 54 mm in May; http://qingdao.tianqi.com/laoshan/). Both CH and CL whole dichotomous inflorescences were collected around 9:00 am −10:00 am, with each inflorescence including 3–5 flower buds at different developmental stages. The first flower bud of a dichotomous inflorescence (the oldest one) is well-developed with all of floral organs about 5 mm in size in the CH flower bud and 3 mm in the CL flower bud. The second flower bud is younger than the first and the third flower bud is the youngest of all. In most cases, there are 1–2 barely-visible flower buds located at the bottom of bract. CH and CL flower buds were collected fresh, frozen immediately in liquid nitrogen and then were transported in dry ice to a freezer where they were stored at −80 °C.
Sequencing
CH and CL flower libraries were generated from flower buds, and each flower bud sample consisted of a mixture of flower buds from different developmental stages obtained from 50 individuals of the same P. heterophylla population. For each kind of flower, total RNA was isolated from the flower buds using Trizol Reagent (Invitrogen) and treated with DNaseI (Promega) to remove residual contaminating DNA. The quality and integrity of the RNA were determined using an Agilent 2100 Bioanalyzer (Agilent). Two RNA-seq libraries (e.g. CH flowers and CL flowers) were prepared using the Illumina TruSeqRNA Sample Preparation Kit (Illumina) following the manufacturer’s instructions. Two normalized cDNA libraries (CH flowers, CL flowers) were constructed and sequenced using the Illumina HiSeq platform (Shanghai Personal Biotechnology Co. Ltd, China) to generate 100 bp paired-end raw reads.
Assembly and functional annotation
Clean reads were obtained by removing adaptor sequences, low-quality reads, and shorter reads (shorter than 50 bp). Trinity (http://trinityrnaseq.github.io) was used to assemble the quality reads into contigs and transcripts [71]. Basic Local Alignment Search Tool (BLAST) was utilized for searching sequence alignments (E-value <1.00E-5) between unique sequences and the non-redundant NCBI (http://www.ncbi.nlm.nih.gov/) and Swiss-Prot (http://www.expasy.ch/sprot) databases. The best-hit transcripts were selected as unigenes. The unigenes with functional categories were annotated by the Gene Ontology database (GO, http://www.geneontology.org) [72], Non-supervised Orthologous Groups (eggNOG) database (http://eggnog.embl.de/) [73], and the Kyoto Encyclopedia of Genes and Genomes database (KEGG, (http://www.genome.jp/kegg/) [74].
Identification of differentially expressed transcripts
Reads per kilobase of exon per million mapped reads (RPKM) [75] were used to normalize the abundance of transcripts on the basis of eliminating the influence of different gene lengths and sequencing discrepancies. Transcripts differentially expressed between the CH and CL flower libraries were identified using DEseq [76]. DETs between two libraries were required to have an absolute value of a log2foldchange (log2FC) > 2 in expression, a p-value of <0.001and an FDR <0.05 [77, 78].
The DETs were subjected to GO and KEGG Orthology (KO) enrichment analysis on the basis of a hypergeometric test (cutoff: FDR <0.05). The KEGG database was also used to predict the pathways in which the DETs were involved, and the pathway annotation was identified by the KEGG mapper (http://www.genome.jp/kegg/tool/map_pathway2.html).
Identification of genes involved in flowering processes
A total of 599Arabidopsis genes associated with gene regulatory networks controlling flowering processes were obtained from GO terms (GO: 0009908 flower development, GO: 0009909 regulation of flower development, GO: 0048439 flower morphogenesis, GO: 0048438 flower whorl development; GO: 0048440 carpel development, GO: 0010093 specification of floral organ identity, GO: 0048497 maintenance of floral organ identity, etc., http://amigo.geneontology.org/) as well as from Srikanth & Schmid [22] and Alvarez-Buylla et al. [24]. The sequences of these genes were downloaded from the Arabidopsis Information Resource (TAIR, http://Arabidopsis.org/). We used the Arabidopsis genes involved in flowering processes as query sequences to search potential homologous genes among the transcript sequences of P. heterophylla by stand-alone BLAST (2.2.30+ version, ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/) with an E-value <1.00E-10. All hits with identities above 60 % were considered to be homologous genes in P. heterophylla.
Validation of floral genes by quantitative real-time PCR
Twenty-four transcripts related to gene regulatory networks of flowering processes identified in the transcriptome sequencing analysis were validated and quantified using quantitative real-time PCR (qRT-PCR). Primers were designed using Primer Premier 5 according to transcriptome sequencing data (Additional file 4). Total RNA was obtained from the biological replicates of those samples used for RNA sequencing. Reverse cDNA was synthesized using PrimeScript Reagent Kit with gDNA Eraser (Takara), and RT-PCR was performed using an Eppendorf Master cycler. P. heterophylla β-actin was used as an internal control to normalize the expression level, and all experiments were performed with three repeats. The reaction was carried out in a total volume of 10 μL containing 5 μL SYBR Green Master Mix (Takara), 1 μL diluted cDNA mix, 0.5 μL each primer (10 mM), and 3 μL RNase-free water. The reactions were performed in a LightCycler480 real-time PCR system (Roche) at 95 °C for 30 s, followed by 45 cycles of 95 °C for 5 s and 60 °C for 30 s. Amplification and detection of only one PCR product was confirmed by performing melting curve analysis of the amplification products at the end of each PCR. After the PCR program, the expression level of different genes was analysed using the comparative CT method (2-△△CT method) [79].
Availability of supporting data
The sequence datasets supporting the results of this article are available from the Short Read Archive (SRA) database at NCBI under the accession SRP052597.
Abbreviations
- CL:
-
cleistogamous
- CH:
-
chasmogamous
- DETs:
-
Differentially expressed transcripts
- qRT-PCR:
-
quantitative real-time PCR
- BLAST:
-
Basic Local Alignment Search Tool
- GRNs:
-
gene regulatory networks
- FM:
-
flower meristem
References
Lord EM. Cleistogamy: a tool for the study of floral morphogenesis, function and evolution. Bot Rev. 1981;47:421–49.
Plitmann U. Distribution of dimorphic flowers as related to other elements of the reproductive strategy. Plant Species Biol. 1995;10:53–60.
Culley TM, Klooster MR. The cleistogamous breeding system: a review of its frequency, evolution, and ecology in Angiosperms. Bot Rev. 2007;73:1–30.
Minter TC, Lord EM. A comparison of cleistogamous and chasmogamous floral development in Collomia grandiflora Dougl. Ex Lindl. (Polemoniaceae). Am J Bot. 1983;70:1499–508.
Lord EM, Eckard KJ, Crone W. Development of the dimorphic anthers in Collomia grandiflora: evidence for heterochrony in the evolution of the cleistogamous anther. J EvolBiol. 1989;2:81–93.
Zhang Y, Yang J, Rao G-Y. Comparative study on the aerial and subterranean flower development in Amphicarpaea edgeworthii Benth. (Leguminosae: Papilionoideae), an amphicarpic species. Int J Plant Sci. 2006;167:943–9.
Luo Y, Bian F-H, Luo Y-B. Different patterns of floral ontogeny in dimorphic flowers of Pseudostellaria heterophylla (Caryophyllaceae). Int J Plant Sci. 2012;173:150–60.
Bell TJ, Quinn JA. Effects of soil moisture and light intensity on the chasmogamous and cleistogamous components of reproductive effort of Dichanthelium clandestinum populations. Can J Bot. 1987;65:2243–9.
Le Corff J. Effects of light and nutrient availability on chasmogamy and cleistogamy in an understory tropical herb, Calathea micans (Marantaceae). Am J Bot. 1993;80:1392–9.
Mattila T, Salonen V. Reproduction of Viola mirabilis in relation to light and nutrient availability. Can J Bot. 1995;73:1917–24.
Darwin C. The different forms of flowers on plants of the same species. London: John Murray; 1877.
Schemske DW. Evolution of reproductive characteristics in Impatiens (Balsaminaceae): the significance of cleistogamy and chasmogamy. Ecology. 1978;59:596–613.
Waller DM. Differences in fitness between seedlings derived from cleistogamous and chasmogamous flowers in Impatiens capensis. Evolution. 1984;38:427–40.
Ni D-H, Li J, Duan Y-B, Yang Y-C, Wei P-C, Xu R-F, et al. Identification and utilization of cleistogamy gene cl7(t) in rice (Oryza sativa L.). J Exp Bot. 2014;65:2107–17.
Nair SK, Wang N, Turuspekov Y, Pourkheirandish M, Sinsuwongwat S, Chen GX, et al. Cleistogamous flowering in barley arises from the suppression of microRNA-guided HvAP2 mRNA cleavage. Proc Natl Acad Sci. 2010;107:490–5.
Lu Y-H, Arnaud D, Belcram H. A dominant point mutation in a RINGv E3 Ubiquitin Ligase homoeologous gene leads to cleistogamy in Brassica napus. Plant Cell. 2012;24:4875–91.
Clay K. Environmental and genetic determinants of cleistogamy in a natural population of the grass Danthonia spicata. Evolution. 1982;36:734–41.
Morinaga SI, Nagano AJ, Miyazaki S, Kudo M, Demura T, Fukuda H, et al. Ecogenomics of cleistogamous and chasmogamous flowering: genome-wide gene expression patterns from cross-species microarray analysis in Cardamine kokaiensis (Brassicaceae). J Ecol. 2008;96:1086–97.
Ke P. Pseudostellaria. In: Tang CL, editor. Flora Reipublicae Popularis Sinicae, vol. 26. Beijing: Science Press; 1996. p. 66–74.
Lu D-Q, Rabeler PK. Pseudostellaria. In: Wu C-Y, Raven PH, Hong D-Y, editors. Flora of China, vol. 6. Beijing: Science Press, and St. Louis: Missouri Botanical Garden Press; 2001. p. 7–10.
Amasino R. Seasonal and developmental timing of flowering. Plant J. 2010;61:1001–13.
Srikanth A, Schmid M. Regulation of flowering time: all roads lead to Rome. Cell Mol Life Sci. 2011;68:2013–37.
Luo Y, Guo Z-H, Li L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev Biol. 2013;380:133–44.
Alvarez-Buylla ER, Benítez M, Corvera-Poiré A, Cador ÁC, de Folter C, de Buen AG, et al. Flower Development. The Arabidopsis Book. 2010;8, e0127. doi:10.1199/tab.0127.
Irish VF. The flowering of Arabidopsis flower development. Plant J. 2010;61:1014–28.
Causier B, Schwarz-Sommer Z, Davies B. Floral organ identity: 20 years of ABCs. Semin Cell Dev Biol. 2010;21:73–9.
Litt A, Kramer EM. The ABC model and the diversification of floral organ identity. Semin Cell Dev Biol. 2010;21:129–37.
Posé D, Yant L, Schmid M. The end of innocence: flowering networks explode in complexity. Curr Opin Plant Biol. 2012;15:45–50.
Turnbull C. Long-distance regulation of flowering time. J Exp Bot. 2011;62:4399–413.
Imaizumi T, Schultz TF, Harmon FG, Ho LA, Kay SA. FKF1 F-box protein mediates cyclic degradation of a repressor of CONSTANS in Arabidopsis. Science. 2005;309:293–7.
Sawa M, Nusinow DA, Kay SA, Imaizumi T. FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science. 2007;318:261–5.
Salazar JD, Saithong T, Brown PE, Foreman J, Locke JCW, Halliday KJ, et al. Prediction of photoperiodic regulators from quantitative gene circuit models. Cell. 2009;139:1170–9.
Ito S, Song Y-H, Imaizumi T. LOV domain-containing F-Box proteins: light-dependent protein degradation modules in Arabidopsis. Mol Plant. 2012;5:1–10.
Corbesier L, Vincent C, Jang S-H, Fornara F, Fan Q-Z, Searle L, et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science. 2007;316:1030–3.
Tamaki S, Matsuo S, Wong H, Yokoi S, Shimamoto K. Hd3a protein is a mobile flowering signal in rice. Science. 2007;316:1033–6.
Lifschitz E, Eshed Y. Universal florigenic signals triggered by FT homologues regulate growth and flowering cycles in perennial day neutral tomato. J Exp Bot. 2006;57:3405–14.
Lin M-K, Belanger H, Lee Y-J, Varkonyi-Gasic E, Taoka KI, Miura E, et al. FLOWERING LOCUS T protein may act as the long-distance florigenic signal in the cucurbits. Plant Cell. 2007;19:1488–506.
Hayama R, Agashe B, Luley E, King R, Coupland G. A circadian rhythm set by dusk determines the expression of nFT homologs and the short-day photoperiodic flowering response in Pharbitis. Plant Cell. 2007;19:2988–3000.
Searle I, He Y-H, Turck F, Coral V, Fornara F, Krober S, et al. The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 2006;20:898–912.
Zik M, Irish VF. Flower development: initiation, differentiation, and diversification. Annu Rev Cell Dev Biol. 2003;19:119–40.
Irish VF, Sussex IM. Function of the apetela-1 gene during Arabidopsis floral development. Plant Cell. 1990;2:741–53.
Liu C, Thong Z, Yu H. Coming into bloom: the specification of floral meristems. Development. 2009;136:3379–91.
Wigge PA, Kim MC, Jaeger KE, Busch W, Schmid M, Lohmann JU, et al. Integration of spatial and temporal information during floral induction in Arabidopsis. Science. 2005;309:1056–9.
Chujo A, Zhang Z, Kishino H, Shimamoto K, Kyozuka J. Partial conservation of LFY function between rice and Arabidopsis. Plant Cell Physio. 2003;44:1311–9.
Blazquez MA, Ferrandiz C, Madueno F, Parcy F. How floral meristems are built. Plant Mol Biol. 2006;60:855–70.
Wagner D, Sablowski RWM, Meyerowitz EM. Transcriptional activation of APETALA1 by LEAFY. Science. 1999;285:582–4.
Ruiz-García L, Madueńo F, Wilkinson M, Haughn G, Salinas J, Martı’nez-Zapater JM. Different roles of flowering-time genes in the activation of floral initiation genes in Arabidopsis. Plant Cell. 1997;9:1921–34.
Kanrar S, Bhattacharya M, Arthur B, Courtier J, Smith HM. Regulatory networks that function to specify flower meristems require the function of homeobox genes PENNYWISE and POUND-FOOLISH in Arabidopsis. Plant J. 2008;54:924–37.
Long J, Barton MK. Initiation of axillary and floral meristems in Arabidopsis. Dev Biol. 2000;218:341–53.
Smith HMS, Ung N, Lal S, Courtier J. Specification of reproductive meristems requires the combined function of SHOOT MERISTEMLESS and floral integrators FLOWERING LOCUS T and FD during Arabidopsis inflorescence development. J Exp Bot. 2011;62:583–93.
Liljegren SJ, Gustafson-Brown C, Pinyopich A, Ditta GS, Yanofsky MF. Interactions among APETALA1, LEAFY, and TERMINAL FLOWER1 specify meristem fate. Plant Cell. 1999;11:1007–18.
Krizek BA, Fletcher JC. Molecular mechanisms of flower development: an armchair guide. Nat Rev Genet. 2005;6:688–98.
Mandel MA, Gustafson-Brown C, Savidge B, Yanofsky MF. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature. 1992;360:273–7.
Bowman JL, Alvarez J, Weigel D, Meyerowitz EM, Smyth DR. Control of flower development in Arabidopsis thaliana by APETALA1 and interacting genes. Development. 1993;119:721–43.
Theissen G, Becker A, Di Rosa A, Kanno A, Kim JT, Munster T, et al. A short history of MADS-box genes in plants. Plant Mol Biol. 2000;42:115–49.
Whipple C, Ciceri P, Padilla CM, Ambrose BA, Bandong SL, Schmidt RJ. Conservation of B-class floral homeotic gene function between maize and Arabidopsis. Development. 2004;131:6083–91.
Mondragon-Palomino M, Theissen G. Conserved differential expression of paralogous DEFICIENS- and GLOBOSA-like MADS-box genes in the flowers of Orchidaceae: refining the ‘orchid code’. Plant J. 2011;66:1008–19.
Zhang R, Guo C-C, Zhang W-G, Wang P-P, Li L, Duan X-S, et al. Disruption of the petal identity gene APETALA3-3 is highly correlated with loss of petals within the buttercup family (Ranunculaceae). Proc Natl Acad Sci U S A. 2013;110:5074–9.
Wang W, Vinocur B, Shoseyov O, Altman A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004;9:244–52.
Bogeat-Triboulot MB, Brosche M, Renaut J, Jouve L, Le Thiec D, Fayyaz P, et al. Gradual soil water depletion results in reversible changes of gene expression, protein profiles, ecophysiology, and growth performance in Populus euphratica, a poplar growing in arid regions. Plant Physiol. 2007;143:876–92.
Akashi K, Yoshida K, Kuwano M, Kajikawa M, Yoshimura K, Hoshiyasu S, et al. Dynamic changes in the leaf proteome of a C3 xerophyte, Citrullus lanatus(wild watermelon), in response to water deficit. Planta. 2011;233:947–60.
Yang K, Lim C-J, Hong J-K, Jin Z-L, Hong J-C, Yun D-J. Identification of Chinese cabbage genes up-regulated by prolonged cold by using microarray analysis. Plant Sci. 2005;168:955–66.
Rushton RJ, Somssich IE, Patricia Ringler P, Qingxi J, Shen QJ. WRKY transcription factors. Trends Plant Sci. 2010;15:247–58.
Yu Y-C, Hu R-B, Wang H-M, Cao Y-P, Guo H, Fu C-X, et al. MIWRKY12, a novel Miscanthus transcription factor, participates in pith secondary cell wall formation and promotes flowering. Plant Sci. 2013;211:1–9.
Cai Y-H, Chen X-J, Xie K, Xing Q-K, Wu Y-W, Li J, et al. Dlf1, a WRKY transcription factor, is involved in the control of flowering time and plant height in rice. PLoS One. 2014;9, e102529.
Castellarin SD, Gaspero GD, Marconi R, Nonis A, Peterlunger E, Paillard S. Colour variation in red grapevines (Vitis vinifera L.): genomic organisation, expression of flavonoid 3′-hydroxylase, flavonoid 3′,5′-hydroxylase genes and related metabolite profiling of red cyanidin-/blue delphinidin-based anthocyanins in berry skin. BMC Genomics. 2006;7:12.
Nakatsuka T, Nishihara M, Mishiba K, Yamamura S. Temporal expression of flavonoid biosynthesis-related genes regulates flower pigmentation in gentian plants. Plant Sci. 2005;168:1309–18.
Brouillard R, Dangeles O. Flavonoids and flower color. In: Harbrone JB, editor. The Flavonoid, Advances in Research since 1986. London: Chapman & Hall; 1993. p. 565–88.
Tanaka Y, Yonekura K, Fukuchi-Mizutani M, Fukui Y, Fujiwara H, Ashikari T, et al. Molecular and biochemical characterization of three anthocyanin synthetic enzymes from Gentianatriflora. Plant Cell Physiol. 1996;37:711–6.
Bloor AJ. A macrocyclic anthocyanin from red/mauve carnation flowers. Phytochemistry. 1997;49:225–8.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40:D284–9.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res. 2012;40:D109–14.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 11: R106. https://genomebiology.biomedcentral.com/articles/10.1186/gb-2010-11-10-r106.
Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7:986–95.
Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29:1165–88.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using Real-Time Quantitative PCR and the 2-△△CT Method. Methods. 2001;25:402–8.
Acknowledgements
We thank two anonymous reviewers for critical and useful comments on the manuscript. We thank Jian-Ping Deng at Ocean University of China, Mei Liu at Institute of Oceanology, Chinese Academy of Sciences, and Fu-Hua Bian at Yantai University for valuable help in collecting samples. We are grateful to Zeng-Fu Xu, Jiang-Hua Chen at Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, and Larry J. Leamy at University of North Carolina at Charlotte for helpful discussion and proofreading. We thank American Journal Experts for English language editing. The authors gratefully acknowledge the Central Laboratory of the Xishuangbanna Tropical Botanical Garden for providing research facilities. This research was supported by the National Natural Science Foundation of China (31270283) and the CAS 135 Program (XTBG-F01,T01).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YL designed experiments, interpreted the data and wrote the manuscript. BHS and JYH designed experiments, analysed RNA-seq data and manuscript editing. LL collected tissue samples, performed library construction. YLL performed RNA-seq analysis. PFW conducted the qPCR validation experiments. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Distribution of contig lengths (a), transcripts lengths (b) and unigene lengths (c). (PDF 556 kb)
Additional file 2:
Transcripts annotated by GO, KEGG, and eggNOG. (XLSX 4808 kb)
Additional file 3:
Putative homologues of Pseudostellaria heterophylla genes involved in flowering process identified by BLAST search with Arabidopsis genes. (XLSX 56 kb)
Additional file 4:
24 floral genes of quantitative real time PCR for validation of RNA-Seq data. (XLSX 13 kb)
Additional file 5:
Differentially expressed transcripts between CH and CL flowers. In total, 396 genes were identified as differentially expressed (|log2FC| > 2, p-value < 0.001 and FDR < 0.05) between CH and CL, including 93 genes that were up-regulated in CH (pink shadow), and 303 genes that were up-regulated (green shadow) in CL. (XLSX 184 kb)
Additional file 6:
GO Enrichment analysis of differentially expressed transcripts. (XLSX 26 kb)
Additional file 7:
KEGG Enrichment analysis of differentially expressed transcripts. (XLSX 12 kb)
Additional file 8:
KEGG pathways classification for differentially expressed transcripts between CH (A) and CL (B) flowers. Red dot represented transcripts up-regulated in CH flowers, green dot represented transcript up-regulated in CL flowers. (XLSX 36 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Luo, Y., Hu, JY., Li, L. et al. Genome-wide analysis of gene expression reveals gene regulatory networks that regulate chasmogamous and cleistogamous flowering in Pseudostellaria heterophylla (Caryophyllaceae). BMC Genomics 17, 382 (2016). https://doi.org/10.1186/s12864-016-2732-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-2732-0