
Abstract
Background
Research has increasingly highlighted the role of serotonin in behavior. However, few researchers have examined serotonin in an evolutionary context, although such research could provide insight into the evolution of important behaviors. The genus Macaca represents a useful model to address this, as this genus shows a wide range of behavioral variation. In addition, many genetic features of the macaque serotonin system are similar to those of humans, and as common models in biomedical research, knowledge of the genetic variation and evolution of serotonin functioning in macaques are particularly relevant for studies of human evolution. Here, we examine the role of selection in the macaque serotonin system by comparing patterns of genetic variation for two genes that code for two types of serotonin receptors - HTR1A and HTR1B - across five species of macaques.
Results
The pattern of variation is significantly different for HTR1A compared to HTR1B. Specifically, there is an increase in between-species variation compared to within-species variation for HTR1A. Phylogenetic analyses indicate that portions of HTR1A show an elevated level of nonsynonymous substitutions. Together these analyses are indicative of positive selection acting on HTR1A, but not HTR1B. Furthermore, the haplotype network for HTR1A is inconsistent with the species tree, potentially due to both deep coalescence and selection.
Conclusions
The results of this study indicate distinct evolutionary histories for HTR1A and HTR1B, with HTR1A showing evidence of selection and a high level of divergence among species, a factor which may have an impact on biomedical research that uses these species as models. The wide genetic variation of HTR1A may also explain some of the species differences in behavior, although further studies on the phenotypic effect of the sequenced polymorphisms are needed to confirm this.
Similar content being viewed by others
Background
Over the past 50 years, research has increasingly highlighted the role of the serotonin system in shaping behavior (for reviews see [1]-[4]). Genetic, pharmaceutical, and hormonal studies have shown that, across taxa, serotonin influences age at dispersal [5],[6], social behavior [7]-[10], exploratory behavior [11], and aggressive behavior [12]-[16], among others. Within humans, it has been shown to play a role in a number of psychological conditions including alcoholism [17],[18], anxiety [19], depression [20],[21], and schizophrenia [22], and many psychiatric medications work by targeting components of the serotonin system. Thus, a detailed understanding of the serotonin system, including its genetic variation and evolution, has wide reaching implications for both biomedical research and for understanding behavioral evolution (see, for example, [16]). However, despite the large amount of research devoted to this system, it is rarely placed in an evolutionary context.
Macaques (Macaca) represent a useful model for addressing the evolution of the serotonin system. Macaques split from other cercopithecine primates approximately 6-8 million years ago (mya); shortly thereafter they migrated from Africa to Asia (approximately 5.5 mya) where they experienced an adaptive radiation [23]-[25]. There are 19-22 extant species known today [26],[27], and a few extinct species are known from the fossil record [23]. While closely related, these species exhibit diverse social behaviors, with some species showing relaxed hierarchies, low levels of severe aggression, and high rates of reconciliation, and others showing strict hierarchies, high levels of severe aggression, and low rates of reconciliation; other behaviors that vary include age at dispersal, mothering styles, degree of kin affiliation, and social play patterns [28],[29], all of which are likely influenced by serotonin. These differences among species are significantly influenced by phylogeny [28],[29], suggesting that genetic differences in neurological functioning may underlie the behavioral differences in this genus [30]. We therefore know that there have been multiple evolutionary events that have driven the diversification of this genus and we might expect to see evolutionary signatures of these events in the genetics of serotonin and other neurological systems.
Moreover, macaques also share interesting parallels with humans. Specifically, they occupy a wide range of habitats, from snowy temperate zones to tropical forests to urban settings, making macaques - particularly the rhesus macaque (Macaca mulatta) - the most widely distributed of nonhuman primates. It has been hypothesized that the exploitation of a large part of the world by both humans and macaques was the result of the evolution of similar behavioral strategies, such as high between-group aggression [31],[32]. Such behaviors are often related to serotonin functioning [5],[6],[12]-[15] and are hypothesized to comprise a "behavioral syndrome" characteristic of invasive species [33]-[35]. In line with this theory, several similar, though independently evolved, genetic variants related to serotonin functioning have been identified in both rhesus macaques and humans, most notably SLC6A4[36] and MAOA[13], and this has been argued to be the result of similar selective pressures acting on the serotonin system [31]. The repeated recruitment of the same neurological system to produce similar behaviors, what we call here parallel evolution sensu Haldane [37], may explain behavioral similarities across a wide range of animals [38],[39]. Thus, understanding the evolution of the serotonin system in macaques may grant insight into the evolution of this system in humans.
Most studies of the serotonin system have focused on the serotonin transporter gene (SLC6A4), but unfortunately this has led to the neglect of other potentially important genes. Two such genes are HTR1A and HTR1B. These two genes code for serotonin receptor types 1A and 1B, and are located on chromosomes 4 and 6 of the macaque genome, respectively [40]. They are similar in structure, having just one exon (1,269 and 1,173 base pairs (bp) for HTR1A and HTR1B, respectively) and no introns; both receptor types are associated with behavior [11],[41]-[45]. While similar in structure and exhibiting an overlap in function [46], experiments with knockout mice indicate that these two receptors modulate behavior in opposite ways [11],[41], potentially due to the different distributions of the receptors within the brain [47]. Knockout mice lacking HTR1A show increased anxiety and decreased exploratory behavior compared to the wildtype [11]; in contrast, knockout mice lacking HTR1B are more impulsive, less anxious, and more aggressive than the wildtype [41]. Thus, differences occurring in the coding and regulatory regions of these receptor genes may contribute to the diverse behaviors macaque species exhibit today.
Here, we begin to place HTR1A and HTR1B in an evolutionary context by investigating the pattern of genetic variation of these genes within and among several species of macaques and testing for the role of selection in these species. By using this approach, we hope to identify the targets of selection, including potentially important mutations that separate species and may contribute to the diversity of macaque behavior. However, as a strictly genetic study, this is merely a first step in examining the evolution of serotonin in macaques; further studies would include directly examining the phenotypic effects of these mutations.
Results
Molecular diversity
Overall, indices of genetic diversity are comparable to those reported for other areas of the macaque genome (Tables 1 and 2; see also Additional file 1: Table S5) [48]. Several studies have found a major split in the M. mulatta lineage between Chinese and Indian populations [24],[49]-[53]. Because the presence of population substructure might have an effect on analyses, we examined representatives of each branch of this species separately. The values for all genetic diversity indices (Tables 1 and 2) are very similar for both lineages, and for all analyses separate examination of the two M. mulatta branches did not affect results. Therefore, for the remainder of the paper, we only report the results for the species as a whole.
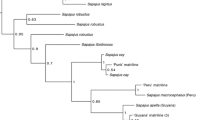
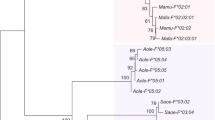
A comparison of the two genes shows that genetic distances between species for HTR1A are generally high compared to HTR1B (Table 3). The only exception to this is between M. mulatta and M. fuscata, where the divergence is actually lower in HTR1A than HTR1B. These trends are also reflected in the gene trees (Figure 1, which includes both coding and noncoding regions; for coding region only, see the Additional file 1: Figure S1) by both the wider spacing of the haplotypes in HTR1A than in HTR1B and the tight clustering of M. mulatta, M. fuscata, and M. cyclopis in HTR1A. Because these three species are members of a monophyletic group that excludes the other macaque species studied (Figure 2), for ease of future discussion, M. mulatta, M. fuscata, and M. cyclopis will be referred to as the mulatta group (see [24],[49],[50]).

Shortest unrooted haplotype networks for HTR1A (left) and HTR1B (right). The networks represent all areas sequenced: coding regions and the areas flanking either side of the gene, including potential regulatory regions. Each circle represents a haplotype whose size is proportional to the frequency of the haplotype. The lengths of the lines connecting the circles are proportional (according to the scale provided) to the number of mutations that separate each haplotype. Because of the larger number of mutations separating the vervet from the macaques in HTR1B, this line (which represent 52 mutations) is not drawn to scale. Sulawesi species include M. nigra, M. tonkeana, and M. maura. For HTR1B, only the original M. nigra is shown (see Methods and Results).

Phylogeny used to run analyses in PAML, which is based on a number of previous molecular studies[62]. The dashed lines indicate the species in which we found both the loss of a codon and two identical nonsynonymous mutations. Based on this phylogeny, these mutations are found in two distinct lineages - the mulatta group and the Sulawesi group (of which M. nigra is a representative) - and is likely a result of deep coalescence.
While members of the same species tend to cluster together in the HTR1A haplotype network, one M. fascicularis individual provides an exception to this pattern by showing greater similarity to a clade with M. nemestrina, M. assamensis, and M. silenus individuals than to the other M. fascicularis individuals (Figure 1). It shares none of the SNPs that distinguish the other M. fascicularis individuals and possesses several SNPs that are unique to this individual. We repeated sequencing on this individual in order to rule out a PCR or sequencing error. This individual likely represents either substructure within M. fascicularis or introgression. It was not clear how this individual would affect our results, so we report here the results of analyses of HTR1A with and without this outlier (Tables 1, 3, and 4). With the exception of the HKA test, inclusion or exclusion of the outlier did not significantly change the test results. Because this individual was largely undifferentiated in HTR1B, its inclusion or exclusion did not significantly alter the results. Therefore, for HTR1B, we only report the results of the original dataset, which includes the outlier.
Evolutionary analyses
For our first set of tests, we applied the HKA test using pairwise comparisons for all species for which we had multiple samples (see Methods). This test compared the levels of within-species variation to between species variation across several loci (see Methods and the Additional file 1: Table S3 for a list of the loci used). We obtained significant results for some between-species comparisons of HTR1A, but not for any comparisons of HTR1B (Table 4). These results were mainly driven by a high level of between-species divergence in HTR1A when compared to the additional loci sequenced (Figure 1; see also Additional file 1: Table S4), and are suggestive of positive selection occurring in macaques. HKA results were not significant for any comparison with M. sylvanus, nor for the comparison between M. mulatta and M. fuscata. All other between-species comparisons are significant for HTR1A when the outlying M. fascicularis individual is excluded. When the outlier is included, only comparisons between M. nemestrina and the mulatta group (M. mulatta and M. fuscata) remain significant. Repeating these analyses using only the coding region of the gene largely replicated these results (Additional file 1: Table S6), although comparison between the mulatta group and M. sylvanus became significant. Likewise, for HTR1B, for some comparisons with M. sylvanus the HKA results for the coding region alone were significant, although in this case this was due to higher than expected diversity within M. sylvanus. For comparisons with Chlorocebus aethiops, see the Additional file 1: Table S7 and Further Discussion.
Our second set of tests focused on the ratio of synonymous to nonsynonymous substitutions (ω). The overall ratios for both genes (Table 4) were almost all either significantly lower than one or else approaching significance. This is consistent with previous studies that have shown the serotonin system to be evolutionarily conserved [54],[55]. However, an examination of ω alone is known to be a highly conservative test, particularly if only a small portion of the entire gene is under selection (see Methods). Therefore, we used phylogenetic analyses using maximum likelihood (PAML), which assigns likelihood values to different models of evolution. Models that incorporate positive selection can be compared to null models and a likelihood ratio test (LRT) used to determine which model best fits the data. Two types of models were compared: variable sites and variable branch. In the variable sites models, ω was allowed to vary among sites; the null models (M1a, M7, and M8a) did not allow for any site to have ω > 1, whereas the selection models (M2a and M8) did. The LRT showed significant results for HTR1A (p <0.001 for all tests), but not for HTR1B (p >0.1 for all tests; Table 5). Bayes empirical Bayes analyses for both M2a and M8 classified codon 15 in HTR1A as a site where ω >1.0 with a high posterior probability (99.2%), indicating that this site of HTR1A was possibly under selection even though the overall ω was low. Two other sites approached significance: codon 22 (with posterior probability of 93.8 and 94.5% for M2a and M8, respectively) and codon 329 (posterior probability of 93.6 and 94.4%). The variable branch models tested to see whether ω varied among branches. The LRT using the variable branch models were non-significant for both genes (p >0.1 for all tests; not shown); that is, no selection was detected on one specific lineage.
Finally, we calculated the Tajima’s D value for both HTR1A and HTR1B, which are shown in Tables 1 and 2. The Tajima’s D values were non-significant (p >0.1) for both genes for all species analyzed.
The placement of M. nigra
Although not closely related to the mulatta group, the haplotype network of HTR1A shows that M. nigra clusters with these species, sharing several derived mutations unique to the mulatta group (Figures 1 and 2). Notably, all four species - M. mulatta, M. fuscata, M. cyclopis, and M. nigra - share both a codon loss (codon 33) and two nonsynonymous substitutions (codons 15 and 22) that no other macaque species exhibit. Based on models of the human serotonin receptor [56], all three mutations affect the extracellular amino terminal region of the receptor. The effect of the codon loss is unknown, however, one nonsynonymous substitution is identical to a polymorphism identified in humans, Gly22Ser [57]. While this polymorphism has not been associated with behavior, possibly due to its low frequency in humans [57],[58], it does show a different pharmacological response than the wildtype [59].
Because it is unexpected for both the mulatta group and M. nigra to share these unusual mutations, especially the loss of a codon, to the exclusion of other, more closely related species (e.g., M. nemestrina), for HTR1A we verified these results by sequencing two additional M. nigra individuals, as well as one individual each from M. maura and M. tonkeana, using the same methods outlined below. M. nigra, M. maura, and M. tonkeana are all located on Sulawesi and are closely related to each other [24], and so we collectively refer to these as the Sulawesi species. All four additional samples had the same codon loss and nonsynonymous mutations, and all cluster with the mulatta group (Figure 1).
Recombination
We estimated the recombination rate as C = 0.002 for HTR1A and C <0.001 for HTR1B using the methods of Hey and Wakeley [60]. For HTR1A, these estimates seem to be largely the result of recombination outside the coding region for HTR1A because when we only included the coding region, we estimated C = 0, suggesting no recombination occurring within the coding region for HTR1A (Additional file 1: Figure S1).
Discussion
Although behaviors are complex and no explanation of behavior can be reduced to a single gene or gene product, it is increasingly recognized that genetic variation plays an important role in mediating social behavior [9],[38],[39]. Because evolution is ultimately a genetic process, understanding behavioral evolution entails understanding the evolution of neurological systems and the genes that underlie them. The serotonin system represents one such genetic/neurological system. Its connection to behavior has been well established and may play a role in the evolution of the wide behavioral diversity found in macaques. Here, a window on the evolution of the serotonin system in macaques is provided by examining the evolutionary history of two serotonin related genes: HTR1A and HTR1B.
A comparison of HTR1A and HTR1B and the possible role of selection
Compared to HTR1B, HTR1A interspecific variation is much higher. While all macaque species cluster tightly together in HTR1B, for HTR1A, three main groups of haplotypes can be discerned: one including M. sylvanus, M. nemestrina, M. silenus, and M. assamensis (with the outlying M. fascicularis individual), a second one consisting of M. fascicularis, and a third one that includes the mulatta group and the Sulawesi macaques. The difference between the two genes can also be seen in the nucleotide diversity among species, which is 2.5 to 6.5 times higher in HTR1A than in HTR1B (and in the additional nonfunctional regions sequenced; see the Additional file 1: Table S4). Most of this variation, both within and among species, is found outside of the coding region. An examination of the coding regions alone shows that, although HTR1A still shows more interspecific divergence, its difference with HTR1B is less pronounced.
Regardless, HTR1A and HTR1B appear to have experienced distinct evolutionary histories and our analyses suggest that different levels of selection on these two genes played a role. Overall ω for both genes was low, suggesting that purifying selection is the dominant force acting on both genes. However, phylogenetic analyses that allowed ω to vary among sites within the coding region of the gene provided evidence that positive selection is acting on HTR1A, but not HTR1B. That is, there is evidence that selection is acting to alter the structure of the protein itself, which is consistent with the finding of both a codon loss and two nonsynonymous substitutions in a subset of the macaque species.
The HKA tests also suggest that for HTR1A, but not HTR1B, non-neutral evolution is occurring. When the outlying M. fascicularis individual was removed, several between-species comparisons were significant for HKA due to the high level of between-species divergence in HTR1A compared to within-species variation, consistent with a positive selective sweep [61]. However, the HKA results were not significant for any comparison with M. sylvanus, or between M. mulatta and M. fuscata. M. mulatta and M. fuscata exhibit a low degree of differentiation from each other (Figure 1), due to their close phylogenetic relationship, which explains this non-significant result. M. sylvanus is the outgroup to all Asian macaque species, being an African relic species and the first to diverge from the other macaque species in any phylogenetic analysis [24],[62],[63]. The non-significant results for comparison to M. sylvanus may indicate that the evolutionary events driving the high level of between-species divergence occurred after the macaque radiation diverged from its African congeners and began to spread across Eurasia. Alternatively, the relatively low sample size for this species (2N = 8) could have contributed to this result. If the outlier is included, comparisons with M. fascicularis become non-significant because the estimate of within-species variation for this species increases dramatically. Even with the inclusion of the outlier, however, results remain significant between M. nemestrina and the mulatta group. These results were largely duplicated when considering only the coding region, even with the reduced variation (although see the Additional file 1: Further Discussion). This, in addition to the PAML results, suggests that natural selection has acted on structure of the HTR1A. However, because a large amount of noncoding variation contributes to the high interspecific diversity found in HTR1A, it is also possible that the function of regulatory regions has been altered as well, although this needs to be confirmed with additional studies.
Macaque species are known to differ greatly in behaviors such as aggression, dispersal, mothering style, and exploratory behavior, all of which have been shown to be modified by serotonin. Thus, we might expect to see a similar level of divergence in genes connected to serotonin. HTR1A shows such a divergence and may be affecting these behaviors in macaques. That this divergence seems to be the result of selection provides evidence that interspecific behavioral variation is not entirely constricted by phylogeny or driven by drift. In a previous study, Claw and colleagues [64] provided evidence that positive selection had occurred on the human lineage in two serotonin-related genes, SLC6A4 and HTR2C, as indicated by an elevated rate of divergence from chimpanzees for the regulatory regions of these genes. While these are not the same genes examined here, they are a part of the general serotonin system, likely the broader target of selection, and it is therefore interesting to compare their results to ours. It has been suggested that parallel evolution of behavioral patterns, via the serotonin system, is occurring between humans and macaques [31] and potentially allowed both taxa to become widespread and adapt to a range of habitats [31],[32]. While we do not address this directly, our results are consistent with this hypothesis and the evidence of positive selection in both taxa is intriguing and warrants further study.
Non-selective evolutionary forces
Although all of the tests used here were designed to detect departures from non-neutral evolution, there are non-selective forces, such as demography and recombination, that may influence these tests; however, these factors seem unlikely to explain the overall results. For example, while demographic events may affect the HKA test [61], this test has also been argued to be somewhat robust to demographic assumptions [65] as demography is expected to have a similar effect on all loci and the HKA test employs a multilocus approach. In addition, Tajima’s D, which is known to be highly sensitive to demography [66], was not significant. That is, it is unclear why a past demographic event might be expected to affect only HTR1A, to the exclusion of HTR1B and the five nonfunctional loci, and why it would significantly affect the HKA test and not Tajima’s D. Furthermore, a demographic event is unlikely to produce the significant results found in PAML, as tests based on ω do not depend on demographic assumptions [61].
Another potential limitation of these analyses is that they are based on captive studies. In particular, the HKA is based on measures of within-species variation, and our estimate might be underestimated by using captive animals. This might be thought of as a specific example of a demographic effect as these populations have (artificially) undergone a bottleneck. We addressed this possibility by selecting nonrelatives from multiple geographic origins (see Methods) in an attempt to capture the true population parameter. However, even if the study design failed to capture the true extent of intraspecific genetic variation, as with other demographic effects, the HKA test should be robust to this problem as the use of captive animals should reduce estimation of within-species variation across all loci.
We did find evidence for recombination, which can affect analyses. This is a conservative assumption for the HKA test, such that the presence of recombination makes it less likely for it to detect selection. However, its presence can lead to an increase in false positives for the PAML comparing various sites models [67]. However, two factors make it unlikely that recombination caused the significant results. First, the tests using PAML are based on the coding region alone, for which we found no recombination. Second, our estimates of C for the entire region were low (C = 0.002). Anisimova et al. [67] showed that a LRT comparing M7 to M8 showed no difference in the rate of false positives when C = 0.001 than when C = 0. While our estimate of recombination is higher than this, our p-values (p <0.001) were well below a significance level of 5%. Nevertheless, we cannot entirely rule out the possibility that non-selective forces are responsible for the variation found in HTR1A.
Population substructure and incongruent gene trees
While the results of the evolutionary analyses are interesting and suggestive of the importance of HTR1A, several other features of HTR1A warrant further discussion, in particular the unusual placement of species and individuals within the haplotype tree. The first is the outlying M. fascicularis individual. Despite there being evidence of recombination occurring around the gene, this individual did not share any of the polymorphisms that distinguished the other M. fascicularis samples and instead showed greater similarity to the M. nemestrina, M. assamensis, and M. silenus samples. This may be the result of either substructure within M. fascicularis or introgression. If the outlier is due to introgression, then this M. fascicularis individual is not representative of the entire M. fascicularis species, and removal of the outlier from analyses is warranted. However, if the individual outlier is due to population substructure, this suggests that the level of within-species genetic variation for HTR1A is extremely high in M. fascicularis compared to other species, which could be the result of balancing selection. Two substitutions that separate the outlier from the rest of the M. fascicularis individuals are a G/C and a C/A mutation at positions 985 and 986 of the exon, respectively, resulting in the substitution of Ala for His at amino acid 329, which is part of the third intracellular loop of the receptor [56]. Mutations in this part of the receptor are known to affect transductional properties [68], thus the genetic variation present in M. fascicularis may be impacting phenotype. What is needed is a broader sampling of M. fascicularis, from a variety of geographical regions, to address the possibility of substructure and balancing selection.
The second unusual result indicated by the haplotype tree of HTR1A is the placement of the Sulawesi macaques with the mulatta group, which includes M. mulatta, M. fuscata, and M. cyclopis, despite the fact that these species are not closely related (see Figure 2). Furthermore, these two groups of species share several derived features not seen in the other macaque species or the vervet, including a complete codon loss and two nonsynonymous mutations, which may have an impact on the receptors’ functionality. When a gene tree is incongruent with a species tree, there are two common explanations. The first is introgression. This seems unlikely based on the geographic distribution of the mulatta species and the Sulawesi species. Furthermore, previous studies examining mitochondrial, Y-chromosomal, and autosomal DNA have not shown evidence for introgression between these two groups [24],[50]. The second explanation, deep coalescence, is more probable. Deep coalescence occurs when the genetic variation that exists between species was present in the ancestral population. While the processes guiding the subsequent sorting of genetic variants among lineages are often thought of as random, deep coalescence creates a situation that allows for parallel evolution. In particular, if species that are not closely related are subjected to similar selective pressures and share genetic variation due to deep coalescence, this would create incongruous trees for the genes under selection.
Because our results suggest that natural selection played a significant role in evolutionary history of HTR1A, it is possible that the placement of the Sulawesi species with the mulatta species is in fact a product of those selection processes. Unfortunately, until future studies can determine the phenotypic effect of the polymorphisms identified here, the exact cause of the genetic variation of HTR1A remains speculative. The grouping of the Sulawesi macaques with the mulatta group might suggest that they experienced similar selective pressures with regard to the serotonin system. However, these groups are quite distinct both behaviorally and ecologically [28],[69]. Studies on the serotonin system have shown that it is connected with behaviors characteristic of invasive species [5]-[15],[33]-[35] and it has been argued that genetic variation in serotonin functioning may allow a species to exploit more habitats [31], potentially through increased plasticity [70],[71]. Outside of humans, M. mulatta has the widest geographic distribution of all primates and their fossil record indicates a long history of expansion [23]. In a similar manner, the geographic location of the Sulawesi macaques are of interest as they have crossed the Makassar Straits between Borneo and Sulawesi and thus Wallace's line at least once [72], something that very few mammalian species have managed to do. Nevertheless, this explanation is also problematic as both M. nemestrina and M. fascicularis are also adept dispersers [73],[74] and yet do not group with the Sulawesi macaques or the mulatta group, and as of yet there is no direct evidence to support this hypothesis. Thus, although our results indicate an unusual evolutionary history for HTR1A, at this stage it is unclear as to its exact cause.
Conclusions
The pattern of variation found in the two genes examined, HTR1A and HTR1B, show that each gene was influenced by different evolutionary forces and that, for HTR1A, positive selection seemed to have played an important role. In particular, two independent lineages of macaques, the mulatta group and the Sulawesi group, show remarkable similarity to each other for HTR1A, to the exclusion of more closely related species, which may be the result of having experienced similar selective pressures. Overall, selection on HTR1A has led to a high level of divergence among species, and this may explain some of the species differences in behavior, including aggression, age at dispersal, and exploratory behavior, although further studies on the phenotypic effect of the sequenced polymorphisms are needed to confirm this. It has been hypothesized that parallel evolution has acted on the serotonin system of both macaques and humans to allow both taxa to successfully exploit a diverse set of habitats over large regions of the world. Because previous research has shown that selection has acted on the serotonin system in humans as well, this study provides support for this hypothesis and opens the door for future studies examining the interaction between genetics, serotonin, and behavior.
Methods
Subjects
We used aliquots of previously extracted DNA from 20 Macaca mulatta (11 from India, 9 from China), 11 M. fascicularis, 11 M. fuscata, 6 M. nemestrina, and 4 M. sylvanus individuals (Additional file 1: Table S1). Previous studies [24],[49]-[53] have shown that M. mulatta consists of two groups that are genetically distinct, roughly split between those of Indian and Chinese origin. Because the substructure of this species could influence our analyses, we analyzed each population separately as well as the species as a whole. In addition to the previously mentioned samples, one sample each from Macaca assamensis, M. cyclopis, M. nigra, M. silenus and Chlorocebus aethiops (Additional file 1: Table S1) were used in PAML analyses (see below) to help place the results in a phylogenetic framework. Because of unusual results obtained for M. nigra for HTR1A, for post hoc analyses on HTR1A we obtained 2 additional M. nigra samples, 1 M. maura, and 1 M. tonkeana. In total, 61 individuals were used producing 122 (=2N) haplotypes. The 11 different macaque species analyzed span the range of behavioral variation found in the genus. The C. aethiops (vervet) sample was used as an outgroup for the macaque species.
The individuals used in this study have been used in previous research and more detailed information can be found in [51],[53],[75]. All primates providing samples for this study were managed in accordance with the United States Public Health Service Policy on the Humane Care and Use of Laboratory Animals. Efforts were made to select nonrelatives that originated from a variety of geographic regions; these decisions were based on breeding records or single-nucleotide polymorphism (SNP)/short tandem repeat analyses [53]. Samples that indicated possible hybridization were avoided, including between Chinese and Indian populations of M. mulatta.
PCR and sequencing
In total, we sequenced 3,119 bp for HTR1A and 2,117 bp for HTR1B. For both genes, we amplified the entire coding region as well as the flanking regions, including potential regulatory regions, using both previously published [76] and newly designed primers (Additional file 1: Table S2). The new primers were designed based on the Macaca mulatta draft assembly [40] using Primer3 [77] and GeneRunner [78]. In addition to HTR1A and HTR1B, five noncoding regions from across the macaque genome were amplified in all of the samples (Additional file 1: Table S3) and used for the HKA test (see below) [79]. These regions have no known function and are thus referred to in this paper as the “nonfunctional regions,” so as to differentiate these regions from the noncoding regions surrounding the serotonin genes, the latter of which may have potential regulatory roles. Because these nonfunctional regions are at least 20,000 base pairs from the nearest coding region [80], they are presumed to be evolving neutrally. The PCR protocols differed for each of the regions amplified and are available upon request. The sequence data for HTR1A and HTR1B are available online [GenBank: KC862384-KC862501]. For the five nonfunctional regions, see [53].
We used ExoSAP-IT to clean up the PCR product and submitted it to the W.M. Keck Center for Comparative and Functional Genomics, UIUC for Sanger sequencing. As with the PCR primers, the sequencing primers used were a combination of published and newly designed primers. Efforts were made to design primers that would provide substantial overlap with each other so that any one region being analyzed would have multiple reads from different primers, ensuring the accuracy and quality of the sequence.
Sequences were aligned and edited manually using Sequencher [81]. Each heterozygote base pair was confirmed visually by identifying clear double peaks in the chromatogram. All SNPs (single nucleotide polymorphisms) and indels (insertions/deletions) were identified. Where indels involved more than one sequential nucleotide, the entire deleted region was treated as a single mutational event. Haplotypes were determined using the program Phase v1 [82]-[84]. In order to visualize the genetic variation and relationships among the haplotypes of each gene, we constructed a haplotype tree using the reduced median method in Network v4.5 (fluxus-engineering.com). All samples sequenced were used to create the tree. Two haplotype trees were created for each gene: one showing the entire region sequenced (both coding and noncoding) and one showing the coding region only.
Analysis
With the exception of the likelihood ratio tests and the haplotype networks, all analyses were conducted on the five species for which we had multiple samples: M. mulatta, M. fuscata, M. fascicularis, M. nemestrina, and M. sylvanus. Several indices of molecular diversity were calculated for each of the five species using Arlequin [85]. These included two different estimates of theta (θ = 4Nμ, where N is the effective population size and θ is the mutation rate): θS[86] and θπ[87], which were used to estimate within-species diversity. Nucleotide diversity (averaged over all loci) [87] was used to estimate within- and between- species diversity.
In order to determine whether selection has acted on either gene, we used multiple approaches. For all selection tests, we used a significance level of 0.05. First, we applied the HKA test, which compares the ratio of within- versus between-species variation in a gene of interest with that of several unlinked, neutral loci. A gene that is evolving neutrally should not have a ratio that varies significantly from that of the other loci considered. We used the five nonfunctional regions described above and in Table S3 (Additional file 1) for comparison to the serotonin genes in the HKA test. In order to get an idea of where selection is occurring, in addition to examining the entire region sequenced for each gene, we also carried out the HKA tests on the coding region alone. Because the HKA test requires a comparison between two species, we conducted pairwise comparisons for all five species using software provided by J. Hey [88]. We also compared each of the five species to the vervet, which is an outgroup to macaques. For each test we ran 10,000 simulations based on parameters estimated from the data to obtain a simulated distribution of the X2 test statistic. P-values were obtained by comparing the observed X2 statistic with the simulated distribution.
Second, we examined the ratio of nonsynonymous to synonymous mutations (ω) in the coding regions of the genes. Positive selection increases the relative rate of nonsynonymous substitutions, whereas purifying selection decreases it [89]. For this approach, we used multiple tests. We used the program Mega v.4 [90] to determine whether ω between any two species significantly differs from 1.0 [89],[91]. If ω >1.0, this indicates positive selection, while ω <1.0 indicates purifying selection. However, this on its own is a very conservative test as there is expected to be a limit to the number and locations of mutations that a gene can maintain while still remaining functional. This means that ω will often be low (ω <1.0) even in the presence of positive selection. Therefore, we conducted likelihood ratio tests (LRT) on all of the samples available using the program PAML (Phylogenetic Analysis using Maximum Likelihood) [92],[93]. PAML determines the maximum likelihood values for different models of evolution within a phylogenetic framework. Models that incorporate positive selection can be compared to null models and the LRT used to determine which model best fits the data. Because PAML requires only one sequence per species, we included all ten species in the LRT using the phylogeny seen in Figure 2[62]. For this study, we examined two categories of evolutionary models. First, we tested various branch models, which allow ω to vary among each of the branches of the macaque phylogeny, to see if selection has occurred on a specific lineage [94]. Tested in this way, ω does not have to be greater than one, but simply elevated compared to other lineages of the macaque phylogeny. Second, we compared several sites models using PAML: M1a vs. M2a, M7 vs. M8, and M8a vs. M8 (see [95]-[97] for detailed explanation of models). These models allow ω to vary among different sites on the gene using different parameters [95],[96],[98]. Most sites on a gene are under strong purifying selection, which reduces the overall ω for a gene and can hide signals of positive selection occurring only in a small portion of the gene. Testing the sites models in PAML can determine if a portion of a gene shows signs of positive selection, even if the average ω ratio over the entire gene is low. Specifically, models M1a, M7, and M8a are null models which assume that there are no class of sites within the gene where ω >1.0 (no positive selection). Models M2a and M8 are similar to the null models except that they allow for an additional category of sites where ω >1.0 (positive selection). For these selection models, a Bayes empirical Bayes test was used to determine the posterior probability that a given site fell into the category where ω >1.0 [97]. A posterior probability of 95% was considered significant.
Finally, using Arlequin, we calculated Tajima’s D for all five species, which detects skews in the frequency spectrum of alleles by comparing two different estimates of theta, θS and θπ[99]. An excess of rare alleles (θS > θπ, leading to significantly negative Tajima's D) is consistent with positive selection, while an excess of intermediate-frequency alleles (θS < θπ, leading to significantly positive Tajima's D) is consistent with balancing selection.
Many of the tests that we employed assume absence of recombination, which can affect the results. Specifically, violation of this assumption can increase false positives in the LRT of the sites models using PAML [67]. In contrast, the assumption of no recombination is conservative for the HKA test [79]. We used the SITES program of J. Hey [88] to obtain an estimate of the recombination parameter C, where C = 2Nc, and c is the rate of recombination per generation per base pair and N is the effective population size [60]. Because the LRT only examines the coding region, whereas the HKA test can analyze both coding and noncoding regions, we obtained estimates of C based on the entire regions sequenced, and on the coding region alone. Presence of recombination indicates that a significant result for the LRT of the sites models should be treated with caution, but should not affect the interpretation of a significant result for HKA.
Additional file
References
Pihl RO, LeMarquand D: Serotonin and aggression and the alcohol-aggression relationship. Alcohol Alcohol. 1998, 33 (1): 55-65. 10.1093/oxfordjournals.alcalc.a008348.
Munafo MR, Clark TG, Moore LR, Payne E, Walton R, Flint J: Genetic polymorphisms and personality in healthy adults: a systematic review and meta-analysis. Mol Psychiatry. 2003, 8 (5): 471-484. 10.1038/sj.mp.4001326.
Cools R, Roberts AC, Robbins TW: Serotoninergic regulation of emotional and behavioural control processes. Trends Cogn Sci. 2008, 12 (1): 31-40. 10.1016/j.tics.2007.10.011.
Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE: Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry. 2010, 167 (5): 509-527. 10.1176/appi.ajp.2010.09101452.
Trefilov A, Berard J, Krawczak M, Schmidtke J: Natal dispersal in rhesus macaques is related to serotonin transporter gene promoter variation. Behav Genet. 2000, 30 (4): 295-301. 10.1023/A:1026597300525.
Krackow S, König B: Microsatellite length polymorphisms associated with dispersal-related agonistic onset in male wild house mice (Mus musculus domesticus). Behav Ecol Sociobiol. 2008, 62 (5): 813-820. 10.1007/s00265-007-0507-y.
Samochowiec J, Lesch KP, Rottmann M, Smolka M, Syagailo YV, Okladnova O, Rommelspacher H, Winterer G, Schmidt LG, Sander T: Association of a regulatory polymorphism in the promoter region of the monoamine oxidase A gene with antisocial alcoholism. Psychiatry Res. 1999, 86 (1): 67-72. 10.1016/S0165-1781(99)00020-7.
Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R: Role of genotype in the cycle of violence in maltreated children. Science. 2002, 297 (5582): 851-854. 10.1126/science.1072290.
Anstey ML, Rogers SM, Ott SR, Burrows M, Simpson SJ: Serotonin mediates behavioral gregarization underlying swarm formation in desert locusts. Science. 2009, 323 (5914): 627-630. 10.1126/science.1165939.
Crockett MJ, Clark L, Hauser MD, Robbins TW: Serotonin selectively influences moral judgment and behavior through effects on harm aversion. Proc Natl Acad Sci U S A. 2010, 107 (40): 17433-17438. 10.1073/pnas.1009396107.
Ramboz S, Oosting R, Amara DA, Kung HF, Blier P, Mendelsohn M, Mann JJ, Brunner D, Hen R: Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc Natl Acad Sci U S A. 1998, 95 (24): 14476-14481. 10.1073/pnas.95.24.14476.
Manuck SB, Flory JD, Ferrell RE, Mann JJ, Muldoon MF: A regulatory polymorphism of the monoamine oxidase-A gene may be associated with variability in aggression, impulsivity, and central nervous system serotonergic responsivity. Psychiatry Res. 2000, 95 (1): 9-23. 10.1016/S0165-1781(00)00162-1.
Newman TK, Syagailo YV, Barr CS, Wendland JR, Champoux M, Graessle M, Suomi SJ, Higley JD, Lesch KP: Monoamine oxidase A gene promoter variation and rearing experience influences aggressive behavior in rhesus monkeys. Biol Psychiatry. 2005, 57 (2): 167-172. 10.1016/j.biopsych.2004.10.012.
Filby AL, Paull GC, Hickmore TF, Tyler CR: Unravelling the neurophysiological basis of aggression in a fish model.BMC Genomics 2010, 11:498.,
Vermeire S, Audenaert K, De Meester R, Vandermeulen E, Waelbers T, De Spiegeleer B, Eersels J, Dobbeleir A, Peremans K: Neuro-imaging the serotonin 2A receptor as a valid biomarker for canine behavioural disorders. Res Vet Sci. 2010, 91 (3): 465-472. 10.1016/j.rvsc.2010.09.021.
Elipot Y, Hinaux H, Callebert J, Retaux S: Evolutionary shift from fighting to foraging in blind cavefish through changes in the serotonin network. Curr Biol. 2013, 23 (1): 1-10. 10.1016/j.cub.2012.10.044.
Virkkunen M, Rawlings R, Tokola R, Poland RE, Guidotti A, Nemeroff C, Bissette G, Kalogeras K, Karonen SL, Linnoila M: CSF biochemistries, glucose metabolism, and diurnal activity rhythms in alcoholic, violent offenders, fire setters, and healthy volunteers. Arch Gen Psychiatry. 1994, 51 (1): 20-27. 10.1001/archpsyc.1994.03950010020003.
Feinn R, Nellissery M, Kranzler HR: Meta-analysis of the association of a functional serotonin transporter promoter polymorphism with alcohol dependence. Am J Med Genet B Neuropsychiatr Genet. 2005, 133B (1): 79-84. 10.1002/ajmg.b.30132.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Muller CR, Hamer DH, Murphy DL: Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996, 274 (5292): 1527-1531. 10.1126/science.274.5292.1527.
Owens MJ, Nemeroff CB: The serotonin transporter and depression. Depress Anxiety. 1998, 8 (Suppl 1): 5-12. 10.1002/(SICI)1520-6394(1998)8:1+<5::AID-DA2>3.0.CO;2-I.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R: Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003, 301 (5631): 386-389. 10.1126/science.1083968.
Meltzer HY, Li Z, Kaneda Y, Ichikawa J: Serotonin receptors: their key role in drugs to treat schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2003, 27 (7): 1159-1172. 10.1016/j.pnpbp.2003.09.010.
Delson E: Fossil macaques, phyletic relationships and a scenario of deployment. The Macaques: Studies in Ecology, Behavior and Evolution. Edited by: Lindburg DG. 1980, Van Nostrand Reinhold, New York, 10-30.
Tosi AJ, Morales JC, Melnick DJ: Paternal, maternal, and biparental molecular markers provide unique windows onto the evolutionary history of macaque monkeys. Evolution. 2003, 57 (6): 1419-1435. 10.1111/j.0014-3820.2003.tb00349.x.
Steiper ME, Young NM: Primate molecular divergence dates. Mol Phylogenet Evol. 2006, 41 (2): 384-394. 10.1016/j.ympev.2006.05.021.
Fooden J: Provisional classification and key to living species of macaques (Primates: Macaca). Folia Primatol. 1976, 25 (2-3): 225-236. 10.1159/000155715.
Fooden J: Classification and distribution of living macaques. The Macaques: Studies in Ecology, Behavior, and Evolution. Edited by: Lindburg DG. 1980, Van Nostrand Reinhold Co, New York, 1-9.
Thierry B, Iwaniuk AN, Pellis SM: The influence of phylogeny on the social behaviour of macaques (Primates: Cercopithecidae, genus Macaca). Ethology. 2000, 106 (8): 713-728. 10.1046/j.1439-0310.2000.00583.x.
Thierry B, Aureli F, Nunn CL, Petit O, Abegg C, de Waal F: A comparative study of conflict resolution in macaques: insights into the nature of trait covariation. Anim Behav. 2008, 75 (3): 847-860. 10.1016/j.anbehav.2007.07.006.
Wendland JR, Lesch KP, Newman TK, Timme A, Gachot-Neveu H, Thierry B, Suomi SJ: Differential functional variability of serotonin transporter and monoamine oxidase A genes in macaque species displaying contrasting levels of aggression-related behavior. Behav Genet. 2006, 36 (2): 163-172. 10.1007/s10519-005-9017-8.
Suomi SJ: Risk, resilience, and gene × environment interactions in rhesus monkeys. Ann N Y Acad Sci. 2006, 1094 (1): 52-62. 10.1196/annals.1376.006.
Maestripieri D: Macachiavellian Intelligence: How Rhesus Macaques and Humans Have Conquered the World. 2007, University of Chicago Press, Chicago, IL
Duckworth RA, Badyaev AV: Coupling of dispersal and aggression facilitates the rapid range expansion of a passerine bird. Proc Natl Acad Sci U S A. 2007, 104 (38): 15017-15022. 10.1073/pnas.0706174104.
Sih A, Bell AM: Insights for behavioral ecology from behavioral syndromes. Adv Study Behav. 2008, 38: 227-281. 10.1016/S0065-3454(08)00005-3.
Cote J, Fogarty S, Weinersmith K, Brodin T, Sih A: Personality traits and dispersal tendency in the invasive mosquitofish (Gambusia affinis). Proc Biol Sci. 2010, 277 (1687): 1571-1579. 10.1098/rspb.2009.2128.
Lesch KP, Meyer J, Glatz K, Flugge G, Hinney A, Hebebrand J, Klauck SM, Poustka A, Poustka F, Bengel D, Mossner R, Riederer P, Heils A: The 5-HT transporter gene-linked polymorphic region (5-HTTLPR) in evolutionary perspective: alternative biallelic variation in rhesus monkeys. J Neural Transm. 1997, 104 (11-12): 1259-1266. 10.1007/BF01294726.
Haldane JBS: The Causes of Evolution. 1932, Longman, London
Fitzpatrick MJ, Ben-Shahar Y, Smid HM, Vet LE, Robinson GE, Sokolowski MB: Candidate genes for behavioural ecology. Trends Ecol Evol. 2005, 20 (2): 96-104. 10.1016/j.tree.2004.11.017.
Robinson GE, Fernald RD, Clayton DF: Genes and social behavior. Science. 2008, 322 (5903): 896-900. 10.1126/science.1159277.
Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007, 316 (5822): 222-234. 10.1126/science.1139247.
Ramboz S, Saudou F, Amara DA, Belzung C, Segu L, Misslin R, Buhot MC, Hen R: 5-HT1B receptor knock out-behavioral consequences. Behav Brain Res. 1996, 73 (1-2): 305-312.
Ramboz S, Saudou F, Amara DA, Belzung C, Dierich A, LeMeur M, Segu L, Misslin R, Buhot MC, Hen R: Behavioral characterization of mice packing the 5-HT1B receptor. NIDA Res Monogr. 1996, 161: 39-57.
Davidge KM, Atkinson L, Douglas L, Lee V, Shapiro S, Kennedy JL, Beitchman JH: Association of the serotonin transporter and 5HT1Dbeta receptor genes with extreme, persistent and pervasive aggressive behaviour in children. Psychiatr Genet. 2004, 14 (3): 143-146. 10.1097/00041444-200409000-00004.
de Boer SF, Koolhaas JM: 5-HT1A and 5-HT1B receptor agonists and aggression: a pharmacological challenge of the serotonin deficiency hypothesis. Eur J Pharmacol. 2005, 526 (1-3): 125-139. 10.1016/j.ejphar.2005.09.065.
Centenaro LA, Vieira K, Zimmermann N, Miczek KA, Lucion AB, de Almeida RM: Social instigation and aggressive behavior in mice: role of 5-HT1A and 5-HT1B receptors in the prefrontal cortex. Psychopharmacol (Berl). 2008, 201 (2): 237-248. 10.1007/s00213-008-1269-6.
Barnes NM, Sharp T: A review of central 5-HT receptors and their function. Neuropharmacology. 1999, 38 (8): 1083-1152. 10.1016/S0028-3908(99)00010-6.
Ghavami A, Stark KL, Jareb M, Ramboz S, Segu L, Hen R: Differential addressing of 5-HT1A and 5-HT1B receptors in epithelial cells and neurons. J Cell Sci. 1999, 112 (Pt 6)(Pt 6): 967-976.
Stevison LS, Kohn MH: Divergence population genetic analysis of hybridization between rhesus and cynomolgus macaques. Mol Ecol. 2009, 18 (11): 2457-2475. 10.1111/j.1365-294X.2009.04212.x.
Melnick DJ, Hoelzer GA, Absher R, Ashley MV: mtDNA diversity in rhesus monkeys reveals overestimates of divergence time and paraphyly with neighboring species. Mol Biol Evol. 1993, 10 (2): 282-295.
Morales JC, Melnick DJ: Phylogenetic relationships of the macaques (Cercopithecidae: Macaca), as revealed by high resolution restriction site mapping of mitochondrial ribosomal genes. J Hum Evol. 1998, 34 (1): 1-23. 10.1006/jhev.1997.0171.
Smith DG: Genetic characterization of Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta). Comp Med. 2005, 55 (3): 227-230.
Hernandez RD, Hubisz MJ, Wheeler DA, Smith DG, Ferguson B, Rogers J, Nazareth L, Indap A, Bourquin T, McPherson J, Muzny D, Gibbs R, Nielsen R, Bustamante CD: Demographic histories and patterns of linkage disequilibrium in Chinese and Indian rhesus macaques. Science. 2007, 316 (5822): 240-243. 10.1126/science.1140462.
Satkoski JA, Malhi RS, Kanthaswamy S, Tito RY, Malladi VS, Smith DG: Pyrosequencing as a method for SNP identification in the rhesus macaque (Macaca mulatta).BMC Genomics 2008, 9:256.,
Andres AM, de Hemptinne C, Bertranpetit J: Heterogeneous rate of protein evolution in serotonin genes. Mol Biol Evol. 2007, 24 (12): 2707-2715. 10.1093/molbev/msm202.
Anbazhagan P, Purushottam M, Kumar HB, Mukherjee O, Jain S, Sowdhamini R: Phylogenetic analysis and selection pressures of 5-HT receptors in human and non-human primates: receptor of an ancient neurotransmitter. J Biomol Struct Dyn. 2010, 27 (5): 581-598. 10.1080/07391102.2010.10508573.
Kobilka BK, Frielle T, Collins S, Yang-Feng T, Kobilka TS, Francke U, Lefkowitz RJ, Caron MG: An intronless gene encoding a potential member of the family of receptors coupled to guanine nucleotide regulatory proteins. Nature. 1987, 329 (6134): 75-79. 10.1038/329075a0.
Nakhai B, Nielsen DA, Linnoila M, Goldman D: Two naturally occurring amino acid substitutions in the human 5-HT1A receptor: glycine 22 to serine 22 and isoleucine 28 to valine 28. Biochem Biophys Res Commun. 1995, 210 (2): 530-536. 10.1006/bbrc.1995.1692.
Bergen A, Wang CY, Nakhai B, Goldman D: Mass allele detection (MAD) of rare 5-HT1A structural variants with allele-specific amplification and electrochemiluminescent detection. Hum Mutat. 1996, 7 (2): 135-143. 10.1002/(SICI)1098-1004(1996)7:2<135::AID-HUMU7>3.0.CO;2-B.
Rotondo A, Nielsen DA, Nakhai B, Hulihan-Giblin B, Bolos A, Goldman D: Agonist-promoted down-regulation and functional desensitization in two naturally occurring variants of the human serotonin1A receptor. Neuropsychopharmacology. 1997, 17 (1): 18-26. 10.1016/S0893-133X(97)00021-3.
Hey J, Wakeley J: A coalescent estimator of the population recombination rate. Genetics. 1997, 145 (3): 833-846.
Nielsen R: Molecular signatures of natural selection. Annu Rev Genet. 2005, 39: 197-218. 10.1146/annurev.genet.39.073003.112420.
Vos RA: Inferring Large Phylogenies: the Big Tree Problem. 2006
Li J, Han K, Xing J, Kim HS, Rogers J, Ryder OA, Disotell T, Yue B, Batzer MA: Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene. 2009, 448 (2): 242-249. 10.1016/j.gene.2009.05.013.
Claw KG, Tito RY, Stone AC, Verrelli BC: Haplotype structure and divergence at human and chimpanzee serotonin transporter and receptor genes: implications for behavioral disorder association analyses. Mol Biol Evol. 2010, 27 (7): 1518-1529. 10.1093/molbev/msq030.
Nachman MW: Detecting selection at the molecular level. Evolutionary Genetics. Edited by: Fox CW, Wolf JB. 2006, Oxford University Press, New York, NY, 103-118.
Tajima F: The effect of change in population size on DNA polymorphism. Genetics. 1989, 123 (3): 597-601.
Anisimova M, Nielsen R, Yang Z: Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003, 164 (3): 1229-1236.
Bruss M, Kostanian A, Bonisch H, Gothert M: The naturally occurring Arg219Leu variant of the human 5-HT1A receptor: impairment of signal transduction. Pharmacogenet Genomics. 2005, 15 (4): 257-264. 10.1097/01213011-200504000-00009.
Thierry B, Bynum E, Baker S, Kinnaird M, Matsumura S, Muroyama Y, O’Brien T, Petit O, Watanabe K: The social repertoire of Sulawesi macaques. Primate Res. 2000, 16: 203-226. 10.2354/psj.16.203.
Belsky J, Jonassaint C, Pluess M, Stanton M, Brummett B, Williams R: Vulnerability genes or plasticity genes?. Mol Psychiatry. 2009, 14 (8): 746-754. 10.1038/mp.2009.44.
Chakraborty S, Chakraborty D, Mukherjee O, Jain S, Ramakrishnan U, Sinha A: Genetic polymorphism in the serotonin transporter promoter region and ecological success in macaques. Behav Genet. 2010, 40 (5): 672-679. 10.1007/s10519-010-9360-2.
Evans BJ, Morales JC, Supriatna J, Melnick DJ: Origin of the Sulawesi macaques (Cercopithecidae: Macaca) as suggested by mitochondrial DNA phylogeny. Biol J Linn Soc. 1999, 66 (4): 539-560.
Fooden J: Ecogeographic segregation of macaque species. Primates. 1982, 23 (4): 574-579. 10.1007/BF02373969.
Abegg C, Thierry B: Macaque evolution and dispersal in insular south-east Asia. Biol J Linn Soc. 2002, 75 (4): 555-576. 10.1046/j.1095-8312.2002.00045.x.
Smith DG, Mcdonough JW, George DA: Mitochondrial DNA variation within and among regional populations of longtail macaques (Macaca fascicularis) in relation to other species of the fascicularis group of macaques. Am J Primatol. 2007, 69 (2): 182-198. 10.1002/ajp.20337.
Cigler T, LaForge KS, McHugh PF, Kapadia SU, Leal SM, Kreek MJ: Novel and previously reported single-nucleotide polymorphisms in the human 5-HT1B receptor gene: No association with cocaine or alcohol abuse or dependence. Am J Med Genet. 2001, 105 (6): 489-497. 10.1002/ajmg.1473.
Rozen S, Skaletsky H: Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000, 132: 365-386.
Gene Runner for Windows [], [http://generunner.net/]
Hudson RR, Kreitman M, Aguade M: A test of neutral molecular evolution based on nucleotide data. Genetics. 1987, 116 (1): 153-159.
Satkoski-Trask JAS, Malhi RS, Kanthaswamy S, Johnson J, Garnica SW, Malladi VS, Smith DG: The influence of SNP discovery method and sample size on the estimation of population genetic parameters in Chinese and Indian rhesus macaques (Macaca mulatta). Primates. 2011, 52: 129-138. 10.1007/s10329-010-0232-4.
Sequencher sequence analysis software [], [www.genecodes.com]
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001, 68 (4): 978-989. 10.1086/319501.
Li N, Stephens M: Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003, 165 (4): 2213-2233.
Stephens M, Donnelly P: A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003, 73 (5): 1162-1169. 10.1086/379378.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005, 1: 47-50.
Watterson GA: On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975, 7 (2): 256-276. 10.1016/0040-5809(75)90020-9.
Tajima F: Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983, 105 (2): 437-460.
Hey J: Heylab software [], [https://bio.cst.temple.edu/~hey/software/software.htm]
Kimura M: Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature. 1977, 267 (5608): 275-276. 10.1038/267275a0.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24 (8): 1596-1599. 10.1093/molbev/msm092.
Yang Z, Nielsen R: Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000, 17 (1): 32-43. 10.1093/oxfordjournals.molbev.a026236.
Yang Z: PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997, 13 (5): 555-556.
Yang Z: PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007, 24 (8): 1586-1591. 10.1093/molbev/msm088.
Yang Z: Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998, 15 (5): 568-573. 10.1093/oxfordjournals.molbev.a025957.
Yang Z, Nielsen R, Goldman N, Pedersen AM: Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000, 155 (1): 431-449.
Swanson WJ, Nielsen R, Yang Q: Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 2003, 20 (1): 18-20. 10.1093/oxfordjournals.molbev.a004233.
Yang Z, Wong WS, Nielsen R: Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005, 22 (4): 1107-1118. 10.1093/molbev/msi097.
Nielsen R, Yang Z: Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998, 148 (3): 929-936.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123 (3): 585-595.
Acknowledgments
We would like to thank S. Williams, R. Neilsen, A. Bell, K. Milich, M. Grabowski, C. Roseman, L. Konigsberg, S. Leigh, and P. Jelinek, along with three anonymous reviewers for helpful feedback on the manuscript. This research was funded by a grant from the National Science Foundation (BCS-0925458) and a portion of the samples provided by the National Non-Human Primate DNA Bank (NIH grant RR000163).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MRS carried out the lab work for the two serotonin genes and performed the analyses of the genetic data. RSM helped to design the study and oversaw all of the lab work and analyses. RYT also contributed to the design of the study and sequenced and aligned the nonfunctional regions. JST, DGS, AD, and DJM provided the DNA samples used in this project and provided input on analyses and interpretation. MRS drafted the initial manuscript and the remaining authors provided feedback for all subsequent revisions. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2014_116_MOESM1_ESM.doc
Additional file 1: Figure S1.: Haplotype networks for the coding regions only of HTR1A (top) and HTR1B (bottom). Table S1. List of sources for species samples. Table S2. List of primers used to sequence HTR1A and HTR1B. Table S3. Additional noncoding loci sequenced. Table S4. Genetic distance within and between species for the five nonfunctional regions sequenced. Table S5. Indices of within-species genetic diversity found in the five nonfunctional regions sequenced. Table S6. Results of the HKA test using only the coding region for HTR1A. Table S7. Results of the HKA test when using Chlorocebus aethiops as the comparative group. Further Discussion. (DOC 462 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Shattuck, M.R., Satkoski-Trask, J., Deinard, A. et al. Patterns of genetic variation and the role of selection in HTR1A and HTR1B in macaques (Macaca). BMC Genet 15, 116 (2014). https://doi.org/10.1186/s12863-014-0116-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-014-0116-5