
Abstract
Background
Addressing genetic issues in the management of fragmented wild populations of threatened species is one of the most important challenges in conservation biology. Nowadays, a diverse array of molecular methods exists to assess genetic diversity and differentiation of wild populations such as allozymes, dominant markers and co-dominant markers. However it remains worthwhile i) to compare the genetic estimates obtained using those several markers in order to ii) test their relative utility, reliability and relevance and iii) the impact of these results for the design of species-specific conservation measures.
Results
Following the successful isolation of 15 microsatellites loci for the cranberry fritillary butterfly, Boloria aquilonaris, we analyzed the genetic diversity and structure of eight populations located in four different landscapes, at both the regional and the landscape scales. We confront results based on microsatellites to those obtained using allozymes and RAPDs on the same samples. Genetic population analyses using different molecular markers indicate that the B. aquilonaris populations are characterized by a weak genetic variation, likely due to low effective population size and low dispersal at the regional scale. This results in inbreeding in some populations, which may have detrimental consequences on their long term viability. However, gene flow within landscape is limited but not inexistent, with some long range movements resulting in low or no isolation by distance. Spatial structuring was detected among the most isolated populations.
Conclusions
The use of allozymes and RAPD are of very limited value to determine population structuring at small spatial (i.e. landscape) scales, microsatellites giving much higher estimate resolution. The use of RAPD data is also limited for evidencing inbreeding. However, coarse-grain spatial structure (i.e. regional scale), and gene flow estimates based on RAPD and microsatellites data gave congruent results. At a time with increasing development of new molecular methods and markers, dominant markers may still be worthwhile to consider in organisms for which no genomic information is available, and for which limited resources are available.
Similar content being viewed by others


Background
Addressing genetic issues in the management of fragmented wild populations of threatened species is one of the most difficult challenges in conservation biology [1]. Indeed, there is now compelling evidence that inbreeding and loss of genetic diversity compromise the viability of populations [2]. Extinction risk is increased through loss of genetic diversity by inbreeding and genetic drift, particularly in small, isolated populations. Accordingly, to elaborate sound management plans, it is necessary to delineate population units, to quantify gene flow, to estimate effective population sizes and to relate genetic diversity to individual fitness and population viability. Nowadays, a diverse array of molecular methods exists to obtain information about the genetic diversity and differentiation of wild populations [3],[4]. However it remains worthwhile i) to compare the results obtained using those several methods and ii) hence to test their reliability and utility (e.g. [5]-[7]).
Variants in amino-acid sequences of the same enzyme due to mutation of genes, or allozymes, were one of the first genetic marker systems used in molecular ecology [8]. They are time and cost effective and have been efficiently applied in population genetic and phylogeographic studies [9]. However, they necessitate invasive sampling, might be subject to selection and may not reveal much variation at small spatial scales and on short time intervals, mainly in species characterized by isolated populations and/or small population sizes [10]. This lack of neutrality is suited in the context of evolutionary studies but can be particularly problematic to test population differentiation. Dominant markers (i.e. markers revealing only a single dominant allele [8]), such as RAPDs (Random Amplified Polymorphic DNA), ISSR (Inter-Simple Sequence Repeats) and AFLP’s (Amplified Fragment Length Polymorphism) have demonstrated their utility (e.g. [11],[12]), even in population assignment tests [13]. However, they remain of limited use in conservation studies where genotypic data are necessary to guide management practices aiming at avoiding inbreeding (i.e. where it is necessary to distinguish heterozygous from homozygous individuals). Although RAPD’s studies do not require much DNA, problems of reproducibility resulted in difficulties to publish RAPD based results [14]. Nonetheless, they remain the least expensive population genetic markers for use on species for which no other genomic information is available.
Co-dominant markers, such as RFLPs (Restriction Fragment Length Polymorphism), microsatellites and SNPs (Single-Nucleotide Polymorphism), (i.e. markers for which all the alleles present at a particular locus can be identified [8]) are nowadays the most widely used molecular markers in population genetic and conservation studies [14]. In particular, microsatellites have become popular and versatile, because they are hyper variable, neutral and reproducible molecular markers [15],[16]. However, developing these markers through microsatellite enrichment followed by Sanger sequencing is time consuming and costly. Genotyping errors and difficulties in isolating microsatellite markers can appear in some groups, including butterflies [17], although Lepidoptera are often considered as an ideal model for conservation biology studies. Indeed, they respond rapidly to both habitat change and global warming [18] and they are good indicators of habitat quality [19]. The difficulty of isolating microsatellites in Lepidoptera [17] has been associated to high similarity in flanking regions between different microsatellites within a same species [20] and/or the lack of conserved flanking regions leading to unrepeatable banding patterns [21]. However, next-generation sequencing offers the opportunity for more efficient microsatellite isolation than was previously possible [22],[23]. The combination of a biotin-enrichment protocol and high-throughput pyrosequencing has recently resulted in the successful isolation of microsatellites for several butterfly species, including the cranberry fritillary Boloria aquilonaris[24], a butterfly species of major conservation concern.
The cranberry fritillary is a glacial relict species whose populations are more or less continuously distributed towards the north of its range, whereas western European populations are highly fragmented. Indeed, suitable habitats are restricted to uplands where climatic conditions are cold and wet. Over the past century, drainage and afforestation of peat bogs (mainly by Norway Spruce Picea abies) has resulted in the substantial decline of the species in this part of its distribution range [25], which has subsequently increased the isolation of the remnant populations.
Intensive demographic and ecological work has been carried out on the Cranberry fritillary in Belgium showing i) a demographic decline in small local populations [26], ii) a relatively high amount of dispersing individuals between closely located populations [27] but iii) also some very long distance dispersal movements [28]. Population genetics has also been used to assess effective connectivity between populations [29],[30]. These studies were essentially based on RAPD (Random Amplified Polymorphic DNA) markers, because allozymes revealed very little polymorphism in the study area. However, RAPD’s revealed low population differentiation within the study landscape, which confirmed that populations were functionally connected, as expected from ecological studies. The genetic structure of the populations inferred from the RAPD data set was indeed remarkably congruent with the geographic configuration of the populations within the landscape and the observed movements among populations [29].
Following the successful isolation of 15 microsatellites loci for B. aquilonaris[24], here we analyze the genetic diversity and structure of eight populations located in four different landscapes. Analyses are hence carried out at both the regional scale (i.e. between landscapes) and the landscape scale (i.e. between populations located in one landscape). We then confront results based on microsatellites to those obtained using allozymes and RAPDs on the same samples [29] and discuss the utility and relevance of these three markers. We also discuss the impact of these results for the design of species-specific conservation measures.
Results
Genetic samples
All of the RAPD loci exhibited overall frequencies of band presence of less than 1 - 3/N (where N is the number of individuals sampled), reducing potential bias in the allele frequency estimates stemming from low counts of band absence phenotype [31]. The results of analyses based on biased and unbiased allele frequencies were very similar, therefore, we only report results based on the latter.
For the microsatellites data, Microchecker analyses indicated the presence of null alleles at two loci in Grande Fange and in Mirenne. However, it should be stressed that no population - primer pair had null allele frequencies of more than 0.001. Simulations showed that the bias induced by null alleles is negligible at frequencies below 0.2 [32]. After controlling for false discovery rate, no primer pair - population combinations were in linkage disequilibrium, nor were any primer - population combinations in Hardy-Weinberg disequilibrium. However, global tests at the population level indicated significant deviance from Hardy-Weinberg equilibrium for four populations (Grande Fange, P =0.0293; Mirenne, P =0.0003; Libin, P =0.0200; Grand Passage, P =0.0346). Hardy-Weinberg disequilibrium in Grande Fange and Grand Passage were due to significant heterozygote deficiency (P =0.0173 and P =0.0057 respectively). Significant heterozygote deficiency was also detected in Pisserotte (P =0.0269).
Genetic diversity estimates
The average number of polymorphic loci (P95) was 0.50 ± 0.13 for allozymes, 0.763 ± 0.118 (ranging 0.526-0.895) for RAPD and 0.895 ± 0.098 (ranging 0.692-1) for microsatellites. The average expected heterozygosity (He) was 0.188 ± 0.029 for allozymes, 0.315 ± 0.047 (ranging 0.239-0.388) for RAPD and 0.485 ± 0.079 (ranging 0.294-0.576) for microsatellites. The mean number of alleles per microsatellite locus (A) was 3.103 ± 0.615, the allelic richness (AR) was 1.250 ± 0.171 and 89.5% of microsatellite loci were polymorphic. Significant inbreeding (f) was detected in Grand Passage and Pisserotte populations (f =0.125 and f =0.130, respectively). All values per populations are presented in Table 1.
N e estimates were obtained for the two populations with the largest sample sizes: Mirenne and Grande Fange. Bayesian and LDNe estimates were not significantly different between populations. LD derived estimates of N e had infinite confidence intervals. Therefore, the Ne:Nc ratio was calculated for Bayesian derived estimates only. Ne:Nc ratios were consistently smaller for the Grande Fange population, due to similar N e estimates but high N c estimates (Table 2). No significant bottleneck was detected, except for the Logbiermé population (Wilcoxon test, P =0.012). However, due to the small sample size, the validity of this significance is uncertain.
Structure of the populations
Population differentiation was high and statistically significant for both RAPDs and microsatellites (Table 3). Many population pairs were significantly differentiated for both marker systems, especially at the regional scale (Table 3). Mirenne population was also significantly differentiated from many populations, even from those within the same upland, confirming results from spatial structure analyses. Previously, significant genetic differentiation was observed for 16 out of the 28 population pairs using allozymes [29]. F st using microsatellites data was estimated at 0.129 (SD: 0.018) at regional scale and 0.082 (SD: 0.016) at the landscape scale (i.e. populations of the Tailles upland only). RAPD based F st values were very similar with 0.171 and 0.091 for the regional and landscape scales, respectively.
Spatial genetic structure detected at the regional scale previously identified i) two clusters (namely the Hautes Fagnes upland vs. the other uplands) based on allozymes and ii) three clusters (namely the Recogne upland vs. the Saint Hubert upland vs. both the Hautes Fagnes and Tailles uplands) based on RAPD [29]. The structure based on microsatellite data was similar between the Bayesian and multivariate analyses. Indeed, the lowest BIC value (multivariate analyses) and the highest likelihood lnP(D) (Bayesian analyses) indicated the presence of five genetic clusters with similar individual attribution to the respective clusters. The most distant uplands, namely Recogne and Saint Hubert, formed separate clusters from Tailles upland and the closely situated Hautes Fagnes upland (Figure 1). However, at the landscape scale, the use of microsatellites enabled the detection of a more fine-scale spatial structure compared to RAPD. Indeed, using RAPD, two genetically distinct clusters were detected, while three clusters were detected using microsatellites (Figure 2). For both markers, the Mirenne population formed a separate genetic cluster. In addition, the microsatellite data identified the Logbiermé population as a distinct cluster from the remaining populations of the Tailles upland. The majority of the individuals in this last cluster (7 out of 9) had a high probability of membership (>0.75).

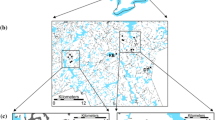
Map of the study system. Black dots: B. aquilonaris populations. Dashed lines: limit of the plateaux. The insert shows the location of the study system in Belgium.

Spatial population structure at the landscape scale (Plateau des Tailles). The probability of membership to hypothetical clusters for a) microsatellite and b) RAPD data.
Dispersal and gene flow
Genetic similarity decreased with increasing distance for both the RAPD (Mantel test: r =0.706, P =0.004) and microsatellite datasets (Mantel test: r =0.599, P =0.022) at the regional scale. It was similar for allozymes, despite not significantly (Mantel test: r =0.414, P =0.092; [29]). However, at the landscape scale (i.e. within the Tailles upland) isolation by distance (IBD) was not significant (PRAPD =0.189, Pmicrosatellite =0.223).
MRR-based estimates of dispersal at the landscape scale were highly correlated to both the effective migration rate estimate (r =0.919, P =0.015) and the maximum likelihood estimate (r =0.912, P =0.019). Using the Akaike information criteria (AIC) in the maximum likelihood analyses of dispersal, the full model (i.e. dispersal between all populations) provided a significantly better fit to the observed genetic data than both stepping stone models. Indeed, the dispersal within 10 km and the between spatial genetic cluster models fitted the data less well (AIC difference with the full model: ΔAIC =18350.17 and ΔAIC =53105.35, respectively). Number of migrants per generation based on the full model were relatively important, with around 50 individual dispersers (Table 4).
Discussion
Genetic diversity in B. aquilonaris populations with DNA markers
Genetic diversity based on microsatellites data measured in B. aquilonaris populations (He =0.485) is low compared to many species, such as the co-occurring specialist butterfly Lycaena helle (He =0.75 in the Pisserotte population; [33],[34]) or as the common butterfly Pararge aegeria (H e =0.828; [34]). Genetic diversity based on RAPD data (He =0.315) was also low and similar to another co-occurring and conservation-targeted species Boloria eunomia (H e =0.368; [35]) and to that of the calcareous grassland specialist butterfly Polyommatus coridon (H e =0.321, based on ISSR dominant markers; [11]). The lowest genetic diversity estimate was given by allozymes (H e =0.188). This low genetic diversity may result from recurrent bottlenecks, a low effective population size N e and/or a genetic isolation of the populations.
Possible bottleneck was detected for only one out of the 8 studied populations. Given the uncertainty of this result (see above), it is hard to conclude that the low genetic diversity emerged or not from recurrent bottlenecks in this case. However, the really low N e and Ne:Nc ratios estimated in the two investigated populations let us think that low N e is one of the source of the genetic paucity for B. aquilonaris. Ne:Nc ratios for B. aquilonaris populations were smaller than the typical average values of the ratio, i.e. between 0.1 and 0.5 [2]. It is although worth mentioning that the high variance in reproductive success typical for invertebrates may result in highly fluctuating Ne:Nc ratios [36]. Therefore, it would be of interest to investigate the temporal fluctuations of the Ne:Nc ratio in this species on large temporal scale. Conversely, Saarinen et al. [37] obtained very high Ne:Nc values (>1.0) in the endangered Miami blue butterfly and attributed this to genetic compensation, i.e. changes in biological interactions at low abundance and hence reduced variance in reproductive success. Higher Ne:Nc in smaller populations was also attributed to genetic compensation in an endangered damselfly [38]. Finally, it should be mentioned that sample size was low in this study, and may explain the large confidence intervals observed for LD estimates of Ne. Initially, destructive sampling was necessary because of associated allozymes studies, therefore limiting the number of samples.
Whatever its origin, genetic paucity is often associated with reduced individual fitness (e.g. [11],[39], but see a contradictory example with Drosophila flies: [40]) and may have negative consequences on the long-term viability of the metapopulation (e.g. [41]). Genetic diversity is also considered as an important asset allowing organism to adapt to climate change, although some confusion still exists as to the relationship between neutral genetic diversity, as measured here, and adaptive genetic diversity. Significant inbreeding was detected in two out of the five populations in the Tailles landscape. Recent population size estimates based on MRR indicate that these two populations decreased dramatically in Nc, while the populations without significant inbreeding did not (Turlure et al. unpublished data).
Gene flow and population structure with DNA markers
Gene flow is important to avoid local inbreeding. Consequently, quantifying gene flow enables landscape management so as to maintain viable metapopulations through recolonization and demographic rescue. Despite high differentiation of the populations, results on DNA markers suggest that some gene flow still occurs between populations of B. aquilonaris, especially within landscapes.
At the regional scale, the degree of genetic differentiation is high, whatever the marker used. Based on the microsatellite data it is very similar to the co-occurring species L. helle[33]. This differentiation is largely influenced by distance, as shown by the significance of the IBD. Note that the IBD was positive but not significant while using allozymes (r =0.414; P =0.092). These results suggest that gene flow is relatively rare at regional scale. This is not surprising as there is not a single population in between the Tailles upland and the Recogne/Saint-Hubert uplands that are separated by c.a. 40 km, which is out of the dispersal range of most specialist butterflies [42].
At the landscape scale, the significance of IBD is lost for both DNA markers. Absence of IBD at relatively small scales has been observed for other butterfly species (e.g. B. eunomia, [43]; and P. coridon, [11]). Absence of IBD at the landscape scale and maximum fit of data to the full migration model together suggest that dispersal is possible between all populations localized within the Tailles upland, and that geographic distance is no longer the principal factor determining the amount of gene flow at this spatial scale. MRR data confirm this, with detection of movements by B. aquilonaris of more than 10 km within the studied landscape ([28]; Turlure unpublished data). Dispersal estimates based on the inverse likelihood function fitted to MRR data and microsatellite data were highly correlated, as it was the case for previously published RAPD data. The ratio of migrants based on demographic estimates versus genetic estimates are respectively 0.765 (SD 1.249) and 0.339 (SD 0.168) for Slatkin’s effective migration rate and the maximum likelihood estimate based on microsatellite data. This indicates that (1) dispersal success is high, with mortality within the matrix remaining limited and (2) immigrants have a high mating success. Spatial analyses with both DNA markers illustrate that the populations of Grande Fange, Grand Passage and Pisserotte are more connected with each other, i.e. they form one genetic cluster, than with the other populations within the Tailles upland. Logbiermé (for microsatellite data only) and especially Mirenne (for both microsatellites and RAPD data) are genetically very different from the other sampled populations. This spatial structure likely results from the geographic isolation of the latter two populations within the Tailles upland.
Implication for B. aquilonaris conservation measures
Genetic population analyses using different molecular markers indicate that the B. aquilonaris populations are characterized by a weak genetic variation, likely due to low effective population size and low dispersal at the regional scale. This results in inbreeding in some populations, which may have detrimental consequences on their long term viability. However, gene flow within landscape is limited but not inexistent, with some long range movements resulting in low or no isolation by distance. Spatial structuring was detected among the most isolated populations. Gene flow between B. aquilonaris populations within landscape is generally superior to one migrant per local population per generation, a genetic rule of thumb to insure the maintenance of genetic diversity and prevent inbreeding depression in fragmented populations [44]. Despite this, significant inbreeding was detected in several populations. Wang [45] suggested that the one migrant per generation rule may be violated when there is variation in size among subpopulations and the number of subpopulations is small. This is the case in this study and it may explain the observed inbreeding despite a number of migrants per generation superior to one. Mills and Allendorf [44] also prescribed more than one migrant per generation when, amongst others, Ne is much less than the total population size. It should be added that too many migrants per generation are not favorable because it may hinders the emergence of local adaptations [46]. In this vein, Mills and Allendorf [44] suggest that up to 10 migrants per generation should not cause uniformity of allele frequencies across subpopulations. In their review, Tallmon et al. [47] highlighted that immigrants may have both positive (i.e. rescue effects) and negative (i.e. outbreeding depression) impacts on the population. Therefore, genetic population analyses of all existing populations on the Tailles upland on multiple generation time series should provide a more detailed picture of gene flow between populations of B. aquilonaris, and is currently underway.
Besides, the conjugation of those results with the knowledge accumulated on the species habitat requirement [48],[49], mobility [50],[51] and dispersal [28] could smoothly guide conservation measures for this species. As the recreation of bog habitat patches in between the current patches is no option, managers may be encouraged 1) to maintain and restore local habitat quality in order to keep suitable census population sizes and 2) to favour the presence of nectar plants along the roads and the rivers as both linear elements are used during dispersal (Turlure, personal observation).
Conclusions
As previously mentioned, allozymes remain of very limited value at small spatial scales; microsatellites giving much higher estimate resolution (e.g. [52]). This is especially true in species such as B. aquilonaris where enzyme variation is extremely limited and hence impedes the correct and accurate estimate of genetic population parameters ([29]; but see contradictory results in [53],[54]). However, here we show that dominant markers may still be worthwhile to consider in organisms for which no genomic information is available, and for which limited (financial and technical) resources are available. Indeed, coarse-grain spatial structure, genetic diversity, and gene flow estimates based on previously published RAPD data gave congruent results with both demographic data and microsatellite-based estimates. Congruent results of population structure and/or differentiation at broad scale while comparing RAPD and microsatellites have also been reported for fishes (e.g. [55]), snakes (e.g. [56],[57]) or ants (e.g. [58]), among other organisms. The use of RAPD data remain however limited for evidencing inbreeding and population structuring at small spatial scale (e.g. [56]). Also, RAPD suffer from poor reproducibility and their use is consequently nowadays limited in ecological studies [59]. New technologies for robust and less expensive genetic polymorphism detection are continuously developed [4], making valuable “new markers” more accessible for population genetics. This is especially possible since the use RAD- and next-generation sequencing generating large amount of DNA sequence or even complete genome sequencing from small amount of genetic material in a relative cost-effective way [60]-[62]. However, their price and accessibility is not always compatible with local conservation issues, as well as the storage and analysis of the volume of sequences produced. Therefore, “old markers”, such as microsatellites or AFLP, may still be valuable markers to answer questions related to species conservation.
Methods
Study system and genetic samples
The cranberry butterfly, Boloria aquilonaris, is a bog specialist butterfly species of conservation interest (listed as ‘vulnerable’ on the Red Data Book of European Butterflies, [63]). It is a glacial relict of acid peat bogs. Adults fly in one generation a year in July and females lay their eggs singly on the host plant Vaccinium oxycoccos. Samples were collected from eight populations in the Belgian Ardenne in 1996, including four different landscapes, namely the uplands of the Hautes-Fagnes, Tailles, Saint-Hubert and Recogne (Figure 1). Samples analysed in this study are identical to those in Vandewoestijne and Baguette [29] with the exception of a few samples for which sufficient DNA was lacking (Table 1).
Genomic DNA was extracted using a phenol-chloroform-isoamyl alcohol method (as described in Vandewoestijne and Baguette [29], followed by RNase treatment. Detection of enzyme polymorphism was carried out as in Vandewoestijne et al. [64]. Only four out of 17 enzyme systems proved to be polymorphic [29]. Due to this extremely low level of polymorphism (e.g. only four polymorphic loci with a mean number of 2.31 alleles), the allozyme data was not re-analyzed in this study; results obtained previously being included for the sake of a broader comparison of markers. Four RAPD primers produced 28 reproducible bands of which 18 were polymorphic. Analyses were carried out as in Vandewoestijne and Baguette [29], i.e. allele frequencies were calculated from null homozygote frequencies assuming panmixia and corrected for dominance according to Lynch & Milligan [31] using TFPGA 1.3 (http://www.marksgeneticsoftware.net/tfpga.htm). Fifteen microsatellite loci were amplified in three multiplex reactions [24]. Multiplex amplifications were conducted using QIAGEN Multiplex PCR Kit (QIAGEN) on Veriti Thermal Cycler (Applied Biosystems) with the following protocol: 95°C for 15 min, followed by 32 cycles (94°C for 30 s, 56°C for 90 s, 72°C for 90 s), and 72°C for 30 min, terminating at 25°C. Allele sizes were scored against the internal standard ROX-400SD (Applied Biosystems) using GeneMapper 3.5® (Applied Biosystems). Hardy Weinberg (HW) equilibrium was tested for all microsatellite primer - population combinations, as well as for each population. Probability values were estimated using the Markov chain method implemented in GENEPOP 4.0.10 (http://genepop.curtin.edu.au/). Linkage disequilibrium was tested for all primer pair - population combinations in GENEPOP using the log likelihood ratio statistic. Significance levels were corrected for false positives (i.e. false discovery rate) following the procedure of Benjamini and Hochberg [65]. In the case of departure from Hardy-Weinberg equilibrium, the data was analysed with Micro-Checker ver. 2.2.0.3 [66]. FREENA [67] was used to calculate null allele frequency whenever detected.
Genetic diversity estimates
Genetic diversity of the sampled populations was characterized through the number of polymorphic loci using the 95% criterion (P95) and unbiased expected heterozygosity (H e ), calculated using TFPGA 1.3 for RAPD data and GENETIX 4.05 (http://www.genetix.univ-montp2.fr/genetix/intro.htm) for microsatellites data. From microsatellites data, we also calculated i) Wright’s inbreeding coefficient f and its significance by permutating alleles within each population (1000 permutations) using GENETIX 4.05, ii) the mean number of alleles per locus (A) using GENETIX 4.05 and iii) the allelic richness (AR) using FSTAT 2.9.3.2 (http://www2.unil.ch/popgen/softwares/fstat.htm).
Effective population size (N e ) was only calculated for populations with a sample size greater than 25 individuals with microsatellites data (as Luikart et al. stated that at least 25 individuals with 10-20 microsatellite loci and 5-10 alleles/locus are required to obtain precise estimates of Ne; [68]). Tallmon et al.’s bayesian computational method was used to estimate N e [69]. This method used multiple summary statistics and hence has improved accuracy and precision compared to other N e estimators because it uses more information from the data [68]. A second point estimator, using linkage disequilibrium information among alleles at different loci caused by genetic drift, LDNe [70] was also used. However, it should be noted that recent simulations have shown at least 60 individuals are necessary to have non biased estimates with the latter estimator [71]. Hence, caution should be used when interpreting LDNe population size estimates. The N e :N c ratio was calculated with estimates of the census population sizes (N c ) in 1995 (i.e. the parental generation). Nc for each population was estimated using Jolly-Seber models fitted on MRR data implemented in the Mark software [72] as in Baguette [28] and following the method described in Schtickzelle et al. [73]. Finally, we tested for a disproportional decrease in allelic diversity compared to heterozygosity due to founder effects following population bottlenecks with BOTTLENECK 1.2.02 [74].
Structure of the populations
Population differentiation for microsatellite loci was estimated using F st sensu Weir & Cockerham [75]. Genotype data was used to derive F st from the variance components (AMOVA) using ARLEQUIN for RAPD data [76].
Discriminant analysis of principal components (DAPC; [77]) and the Bayesian clustering method implemented in STRUCTURE [78] were used to identify and describe clusters of related individuals on both the RAPD and microsatellite data sets. The DAPC method has the advantage that it does not make any assumptions about the underlying population genetic model, although most conservation geneticists are acquainted with, and use the Bayesian clustering method. DAPC analyses were implemented in R 2.12.2 (R Development Core Team 2009) using the adegenet package [79]. A principal component (PCA) was carried out on the genetic data sets. The number of PC’s maintained in the discriminant analyses was determined so as to retain 90% of the genetic variation. The number of clusters was chosen based on the lowest BIC value. STRUCTURE analyses were run with admixture and correlated allele frequencies, 300.000 iterations after an initial burn-in of 100.000 iterations and 10 repetitions for each K. The number of clusters K was determined based on the probability ln P(D)parameter [78]. The second order rate of change of lnP(D) with respect to K [80] was also used to determine the number of clusters using the Structure-sum 2010 package [81] in R 2.12.2 (R Development Core Team 2009). For both clustering approaches, analyses were run on the entire data set to detect the regional structure (i.e. structure between the landscapes) and on populations of the Tailles upland only to detect the landscape structure (i.e. structure within the Tailles upland landscape).
Dispersal and gene flow
In a first step, a Mantel test was used to assess the correlation between the genetic differentiation of the populations (F st as described above) and the geographic distances between populations, at both regional and landscape scales (i.e. under the classical isolation by distance hypothesis, IBD). Geographic distances between populations were calculated as the Euclidian distances between populations’ pairs (using “HawthTool” Analysis Tools in ArcGIS). Significance levels were tested based on 10000 permutations.
In a second step, the correlations between the MRR-based number of migrants and the several genetic-based dispersal estimates were tested through Mantel tests. Dispersal probability was estimated using an inverse power function fitted to Mark Release Recapture (MRR) data as in Vandewoestijne and Baguette [30]. Those MRR data were collected in populations of the Tailles upland only in 1997. Number of migrants based on MRR data was calculated by multiplying the probability of dispersal by the estimated population size. This dispersal measure was compared with several genetic estimates of gene flow. First, the effective migration rate[82] between site pairs was calculated as Fst ≈ 1/(4 Nm +1), where N is the effective population size of each population and m is the migration rate between populations. It should be mentioned that this island model makes a large number of simplifying assumptions. Nonetheless, performance evaluations using both simulations and analytical theory, suggest that the approach gives reasonable estimates of Nm even when certain assumptions are violated [10]. Next, a maximum likelihood estimator based on the coalescent for unequal migration rates and different subpopulation sizes was used [83], using the Brownian motion microsatellite data model, as implemented in MIGRATE v2.1.3. To test which model (stepping stone or island) fitted the observed genetic populations structure the best, the difference (Δi) between their respective AIC value (Akaike Information Criterion) was calculated. Dispersal was possible between all pairs of populations in the full matrix model while, in the stepping stone model, dispersal was only possible between populations separated by less than 10 kilometres. We also tested a second stepping stone model based on the results of spatial genetic analyses, i.e. dispersal was possible between populations within a cluster, but not between clusters. Finally, the number of migrants per generation was calculated as (4Neμ) × (m/μ) where N e is the effective population size, μ the mutation rate and m the immigration rate [83].
References
Frankham R: Challenges and opportunities of genetic approaches to biological conservation. Biol Conserv. 2010, 143: 1919-1927. 10.1016/j.biocon.2010.05.011.
Frankham R: Genetics and extinction. Biol Conserv. 2005, 126: 131-140. 10.1016/j.biocon.2005.05.002.
Arif IA, Khan HA: Molecular markers for biodiversity analysis of wildlife animals: a brief review. Anim Biodivers Conserv. 2009, 32: 9-17.
Behura SK: Molecular marker systems in insects: current trends and future avenues. Mol Ecol. 2006, 15: 3087-3113. 10.1111/j.1365-294X.2006.03014.x.
Zhang DX, Hewitt GM: Nuclear DNA analyses in genetic studies of populations: practice, problems and prospects. Mol Ecol. 2003, 12: 563-584. 10.1046/j.1365-294X.2003.01773.x.
Pogson GH, Mesa KA, Boutilier RG: Genetic population structure and gene flow in the Atlantic cod Gadus morhua: a comparison of allozyme and nuclear RFLP loci. Genetics. 1995, 139: 375-385.
Estoup A, Rousset F, Michalakis Y, Cornuet JM, Adriamanga M, Guyomard R: Comparative analysis of microsatellite and allozyme markers: a case study investigating microgeographic differentiation in brown trout (Salmo trutta). Mol Ecol. 1998, 7: 339-353. 10.1046/j.1365-294X.1998.00362.x.
Freeland J: Molecular Ecology. 2006, John Wiley & Sons, Chichester
Richardson BJ, Baverstock PR, Adams M: Allozyme Electrophoresis: A Handbook for Animal Systematics and Population Studies. 1988, Academic Press, San Diego
Allendorf FW, Luikart G: Conservation and the Genetics of Populations. 2007, Blackwell Publishing, Oxford
Vandewoestijne S, Schtickzelle N, Baguette M: Positive correlation between genetic diversity and fitness in a large, well-connected metapopulation. BMC Biol. 2008, 6: 46-10.1186/1741-7007-6-46.
Meudt HM, Clarke AC: Almost Forgotten or Latest Practice? AFLP applications, analyses and advances. Trends Plant Sci. 2007, 12: 106-117. 10.1016/j.tplants.2007.02.001.
Campbell D, Duchesne P, Bernatchez L: AFLP utility for population assignment studies: analytical investigation and empirical comparison with microsatellites. Mol Ecol. 2003, 12: 1979-1991. 10.1046/j.1365-294X.2003.01856.x.
Finger A, Klank C: Review Molecular Methods: Blessing or Curse?. Relict Species. Edited by: Habel J, Assmann T. 2010, Springer Berlin Heidelberg, Berlin Heidelberg, 309-320. 10.1007/978-3-540-92160-8_18.
Jarne P, Lagoda PJL: Microsatellites, from molecules to populations and back. Trends Ecol Evol. 1996, 11: 424-429. 10.1016/0169-5347(96)10049-5.
Selkoe KA, Toonen RJ: Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett. 2006, 9: 615-629. 10.1111/j.1461-0248.2006.00889.x.
Neve G, Meglecz E: Microsatellite frequencies in different taxa. Trends Ecol Evol. 2010, 15: 376-377. 10.1016/S0169-5347(00)01921-2.
Warren MS, Hill JK, Thomas JA, Asher J, Fox R, Huntley B, Roy DB, Telfer MG, Jeffcoate S, Harding P, Jeffcoate G, Willis SG, Greatorex-Davies JN, Moss D, Thomas CD: Rapid responses of British butterflies to opposing forces of climate and habitat change. Nature. 2001, 414: 65-10.1038/35102054.
New TR: Are Lepidoptera an effective “umbrella group” for biodiversity conservation?. J Insect Conserv. 1997, 1: 5-12. 10.1023/A:1018433406701.
Meglecz E, Petenian F, Danchin E, D'Acier AC, Rasplus JY, Faure E: High similarity between flanking regions of different microsatellites detected within each of two species of Lepidoptera: Parnassius apollo and Euphydryas aurinia. Mol Ecol. 2004, 13: 1693-1700. 10.1111/j.1365-294X.2004.02163.x.
Sarhan A: Isolation and characterization of five microsatellite loci in the Glanville fritillary butterfly (Melitaea cinxia). Mol Ecol Notes. 2006, 6: 163-164. 10.1111/j.1471-8286.2006.01176.x.
Malausa T, Gilles A, Meglecz E, Blanquart H, Duthoy S, Costedoat C, Dubut V, Pech N, Castagnone-Sereno P, Délye C, Feau N, Frey P, Gauthier P, Guillemaud T, Hazard L, Le Corre V, Lung-Escarmant B, Malé P-JG, Ferreira S, Martin J-F: High-throughput microsatellite isolation through 454 GS-FLX Titanium pyrosequencing of enriched DNA libraries. Mol Ecol Resour. 2011, 11: 638-644. 10.1111/j.1755-0998.2011.02992.x.
Abdelkrim J, Robertson BC, Stanton JAL, Gemmel NJ: Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing. Biotechniques. 2009, 46: 185-191. 10.2144/000113084.
Vandewoestijne S, Turlure C, Baguette M: Development of novel microsatellite markers for a specialist species of Lepidoptera, Boloria aquilonaris (Nymphalidae), based on 454 sequences. Eur J Entomol. 2011, 109: 129-134. 10.14411/eje.2012.016.
Fichefet V, Barbier Y, Baugnée J-Y, Dufrêne M, Goffart P, Maes D, Van Dyck H: Papillons de jour de Wallonie (1985-2007). 2008, Publication du Groupe de Travail Papillons de jour Lycaena et du Centre de Recherche de la Nature, des Forêts et du Bois (MRW-DGRNE), Gembloux
Baguette M, Schtickzelle N: Local population dynamics are important to the conservation of metapopulations in highly fragmented landscapes. J Appl Ecol. 2003, 40: 404-412. 10.1046/j.1365-2664.2003.00791.x.
Mousson L, Nève G, Baguette M: Metapopulation structure and conservation of the cranberry fritillary Boloria aquilonaris (Lepidoptera, Nymphalidae) in Belgium. Biol Conserv. 1999, 87: 285-293. 10.1016/S0006-3207(98)00081-0.
Baguette M: Long distance dispersal and landscape occupancy in a metapopulation of the cranberry fritillary butterfly. Ecography. 2003, 26: 153-160. 10.1034/j.1600-0587.2003.03364.x.
Vandewoestijne S, Baguette M: The genetic structure of endangered populations in the Cranberry Fritillary, Boloria aquilonaris (Lepidoptera, Nymphalidae): RAPDs vs allozymes. Heredity. 2002, 89: 439-445. 10.1038/sj.hdy.6800161.
Vandewoestijne S, Baguette M: Demographic versus genetic dispersal measures. Pop Ecol. 2004, 46: 281-285. 10.1007/s10144-004-0197-5.
Lynch M, Milligan BG: Analysis of population genetic structure with RAPD markers. Mol Ecol. 1994, 3: 91-99. 10.1111/j.1365-294X.1994.tb00109.x.
Dakin EE, Avise JC: Microsatellite null alleles in parentage analysis. Heredity. 2004, 93: 504-509. 10.1038/sj.hdy.6800545.
Finger A, Schmitt T, Zachos FE, Meyer M, Assmann T, Habel JC: The genetic status of the violet copper Lycaena helle - a relict of the cold past in times of global warming. Ecography. 2009, 32: 382-390. 10.1111/j.1600-0587.2008.05766.x.
Vandewoestijne S, Van Dyck H: Population genetic differences along a latitudinal cline between original and recently colonized habitat in a butterfly. Plos One. 2010, 5: e13810-10.1371/journal.pone.0013810.
Vandewoestijne S, Baguette M: Genetic population structure of the vulnerable bog fritillary butterfly. Hereditas. 2004, 141: 199-206. 10.1111/j.1601-5223.2004.01849.x.
Hedrick P: Large variance in reproductive success and the Ne/N ratio. Evolution. 2005, 59: 1596-1599. 10.1111/j.0014-3820.2005.tb01809.x.
Saarinen EV, Austin JD, Daniels JC: Genetic estimates of contemporary effective population size in an endangered butterfly indicate a possible role for genetic compensation. Evol Appl. 2010, 3: 28-39. 10.1111/j.1752-4571.2009.00096.x.
Watts PC, Saccheri IJ, Kemp SJ, Thompson DJ: Effective population sizes and migration rates in fragmented populations of an endangered insect (Coenagrion mercuriale: Odonata). J Anim Ecol. 2007, 76: 790-800. 10.1111/j.1365-2656.2007.01249.x.
Reed DH, Frankham R: Correlation between fitness and genetic diversity. Conserv Biol. 2003, 17: 230-237. 10.1046/j.1523-1739.2003.01236.x.
Legrand D, Tenaillon MI, Matyot P, Gerlach J, Lachaise D, Cariou ML: Species-wide genetic variation and demographic history of Drosophila sechellia, a species lacking population structure. Genetics. 2009, 182: 1197-1206. 10.1534/genetics.108.092080.
Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, Hanski I: Inbreeding and extinction in a butterfly metapopulation. Nature. 1998, 392: 491-494. 10.1038/33136.
Stevens VM, Turlure C, Baguette M: A meta-analysis of dispersal in butterflies. Biol Rev. 2010, 85: 625-642.
Nève G, Barascud B, Descimon H, Baguette M: Genetic structure of Proclossiana eunomia populations at the regional scale (Lepidoptera, Nymphalidae). Heredity. 2000, 84: 657-666. 10.1046/j.1365-2540.2000.00699.x.
Mills LS, Allendorf FW: The one-migrant-per-generation rule in conservation and management. Conserv Biol. 1996, 10: 1509-1518. 10.1046/j.1523-1739.1996.10061509.x.
Wang H: Application of the one-migrant-per-generation rule to conservation and management. Conserv Biol. 2004, 18: 332-343. 10.1111/j.1523-1739.2004.00440.x.
Kisdi E: Dispersal: risk spreading versus local adaptation. Am Nat. 2002, 159: 579-596. 10.1086/339989.
Tallmon DA, Luikart G, Waples RS: The alluring simplicity and complex reality of genetic rescue. Trends Ecol Evol. 2004, 19: 489-496. 10.1016/j.tree.2004.07.003.
Turlure C, Choutt J, Baguette M, Van Dyck H: Microclimate buffering and resource-based habitat of a relict butterfly species: significance for conservation under climate change. Global Change Biol. 2010, 16: 1883-1893. 10.1111/j.1365-2486.2009.02133.x.
Turlure C, Choutt J, Van Dyck H, Baguette M, Schtickzelle N: Functional habitat area as a reliable proxy for population size: case study using two butterfly species of conservation concern. J Insect Conserv. 2010, 14: 379-388. 10.1007/s10841-010-9269-3.
Turlure C, Schtickzelle N, Baguette M: Resource grain scales mobility and adult morphology in butterflies. Land Ecol. 2010, 25: 95-108. 10.1007/s10980-009-9403-3.
Turlure C, Baguette M, Stevens VM, Maes D: Species- and sex-specific adjustments of movement behavior to landscape heterogeneity in butterflies. Behav Ecol. 2011, 22: 967-975. 10.1093/beheco/arr077.
Gao L, Schaal B, Zhang C, Jia J, Dong Y: Assessment of population genetic structure in common wild rice Oryza rufipogon Griff. using microsatellite and allozyme markers. Theor Appl Genet. 2002, 106: 173-180.
Freville H, Justy F, Olivieri I: Comparative allozyme and microsatellite population structure in a narrow endemic plant species, Centaurea corymbosa Pourret (Asteraceae). Mol Ecol. 2001, 10: 879-889. 10.1046/j.1365-294X.2001.01249.x.
Allendorf FW, Seeb LW: Concordance of genetic divergence among sockeye salmon populations at allozyme, nuclear DNA, and mitochondrial DNA markers. Evolution. 2000, 54: 640-651. 10.1111/j.0014-3820.2000.tb00065.x.
Abdul Muneer PM, Gopalakrishnan A, Musammilu KK, Mohindra V, Lal KK, Basheer VS, Lakra WS: Genetic variation and population structure of endemic yellow catfish, Horabagrus brachysoma (Bagridae) among three populations of Western Ghat region using RAPD and microsatellite markers. Mol Biol Rep. 2009, 36: 1779-1791. 10.1007/s11033-008-9381-6.
Lougheed SC, Gibbs HL, Prior KA, Weatherhead PJ: A comparison of RAPD versus microsatellite DNA markers in population studies of the Massasauga rattlesnake. J Hered. 2000, 91: 458-463. 10.1093/jhered/91.6.458.
Carlsson M, Söderberg L, Tegelstr m H: The genetic structure of adders (Vipera berus) in Fennoscandia: congruence between different kinds of genetic markers. Mol Ecol. 2004, 13: 3147-3152. 10.1111/j.1365-294X.2004.02315.x.
Ross KG, Shoemaker DD, Krieger MJ, DeHeer CJ, Keller L: Assessing genetic structure with multiple classes of molecular markers: a case study involving the introduced fire ant Solenopsis invicta. Mol Biol Evol. 1999, 16: 525-543. 10.1093/oxfordjournals.molbev.a026134.
Black WC: PCR with arbitrary primers: approach with care. Insect Mol Biol. 1993, 2: 1-6. 10.1111/j.1365-2583.1993.tb00118.x.
Ekblom R, Galindo J: Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011, 107: 1-15. 10.1038/hdy.2010.152.
Allendorf FW, Hohenlohe PA, Luikart G: Genomics and the future of conservation genetics. Nat Rev Genet. 2010, 11: 697-709. 10.1038/nrg2844.
Ouborg NJ, Pertoldi C, Loeschcke V, Bijlsma R, Hedrick PW: Conservation genetics in transition to conservation genomics. Trends Genet. 2010, 26: 177-187. 10.1016/j.tig.2010.01.001.
van Swaay C, Warren M, Lo’s G: Biotope use and trends of European butterflies. J Insect Conserv. 2006, 10: 189-209. 10.1007/s10841-006-6293-4.
Vandewoestijne S, Nève G, Baguette M: Spatial and temporal population genetic structure of the butterfly Aglais urticae L. (Lepidoptera, Nymphalidae). Mol Ecol. 1999, 8: 1539-1543. 10.1046/j.1365-294x.1999.00725.x.
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995, 57: 289-300.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P: MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004, 4: 535-538. 10.1111/j.1471-8286.2004.00684.x.
Chapuis MP, Estoup A: Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007, 24: 621-631. 10.1093/molbev/msl191.
Luikart G, Ryman N, Tallmon D, Schwartz M, Allendorf F: Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet. 2010, 11: 355-373. 10.1007/s10592-010-0050-7.
Tallmon DA, Koyuk A, Luikart G, Beaumont MA: COMPUTER PROGRAMS: onesamp: a program to estimate effective population size using approximate Bayesian computation. Mol Ecol Resour. 2008, 8: 299-301. 10.1111/j.1471-8286.2007.01997.x.
Waples RS, Do C: LDNe: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour. 2008, 8: 753-756. 10.1111/j.1755-0998.2007.02061.x.
Tallmon DA, Gregovich D, Waples RS, Scott Baker C, Jackson J, Taylor BL, Archer E, Martien KK, Allendorf FW, Schwartz MK: When are genetic methods useful for estimating contemporary abundance and detecting population trends?. Mol Ecol Resour. 2010, 10: 684-692. 10.1111/j.1755-0998.2010.02831.x.
White GC, Burnham KP: Program MARK: survival estimation from populations of marked animals. Bird Study. 1999, 46 (Supplement): S120-S139. 10.1080/00063659909477239.
Schtickzelle N, Le Boulengé E, Baguette M: Metapopulation dynamics of the bog fritillary butterfly: demographic processes in a patchy population. Oikos. 2002, 97: 349-360. 10.1034/j.1600-0706.2002.970305.x.
Cornuet JM, Luikart G: Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996, 144: 2001-2014.
Weir BS, Cockerham CC: Estimating F-statistics for the analysis of population structure. Evolution. 1984, 38: 1358-1370. 10.2307/2408641.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform. 2005, 1: 47-50.
Jombart T, Devillard S, Balloux F: Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010, 11: 94-10.1186/1471-2156-11-94.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Jombart T: adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008, 24: 1403-1405. 10.1093/bioinformatics/btn129.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005, 14: 2611-2620. 10.1111/j.1365-294X.2005.02553.x.
Ehrich D: aflpdat: a collection of r functions for convenient handling of AFLP data. Mol Ecol Notes. 2006, 6: 603-604. 10.1111/j.1471-8286.2006.01380.x.
Slatkin M: Isolation by distance in equilibrium and non-equilibrium populations. Evolution. 1993, 47: 264-279. 10.2307/2410134.
Beerli P, Felsenstein J: Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci U S A. 2001, 98: 4563-4568. 10.1073/pnas.081068098.
Acknowledgements
Thanks are due to Linda Dhondt and Christine Noel for help with lab work. We are also grateful to the associate editor and to the reviewers for the thoughtful comments that really improved the quality of the manuscript. SW and CT were post-doctoral fellows of the FRS-FNRS during this research and acknowledge its financial support. This is publication no. BRC324 of the Biodiversity Research Centre of the UCL. MB is part of the Laboratoire d’Excellence TULIP (ANR-10-LABX-41) and acknowledges grants from the ANR (open call INDHET, 6th extinction call MOBIGEN). Site access and a permission to study the species in the field and in the lab were granted by the Ministère de la R gion Wallonne.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SV designed the study, performed data collection and analyses with CT; SV, CT and MB commented, interpreted and participated to the improvement of these analyses. SV and CT drafted the manuscript, and MB contributed substantially to its revisions. All authors read and approved the final version of the paper.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Turlure, C., Vandewoestijne, S. & Baguette, M. Conservation genetics of a threatened butterfly: comparison of allozymes, RAPDs and microsatellites. BMC Genet 15, 114 (2014). https://doi.org/10.1186/s12863-014-0114-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-014-0114-7