
Abstract
Background
Pleistocene climatic oscillations and historical geological events may both influence current patterns of genetic variation, and the species in southern China that faced unique climatic and topographical events have complex evolutionary histories. However, the relative contributions of climatic oscillations and geographical events to the genetic variation of these species remain undetermined. To investigate patterns of genetic variation and to test the hypotheses about the factors that shaped the distribution of this genetic variation in species of southern China, mitochondrial genes (cytochrome b and NADH dehydrogenase subunit 2) and nine microsatellite loci of the Omei tree frog (Rhacophorus omeimontis) were amplified in this study.
Results
The genetic diversity in the populations of R. omeimontis was high. The phylogenetic trees reconstructed from the mitochondrial DNA (mtDNA) haplotypes and the Bayesian genetic clustering analysis based on microsatellite data both revealed that all populations were divided into three lineages (SC, HG and YN). The two most recent splitting events among the lineages coincided with recent geological events (including the intense uplift of the Qinghai-Tibet Plateau, QTP and the subsequent movements of the Yun-Gui Plateau, YGP) and the Pleistocene glaciations. Significant expansion signals were not detected in mismatch analyses or neutrality tests. And the effective population size of each lineage was stable during the Pleistocene.
Conclusions
Based on the results of this study, complex geological events (the recent dramatic uplift of the QTP and the subsequent movements of the YGP) and the Pleistocene glaciations were apparent drivers of the rapid divergence of the R. omeimontis lineages. Each diverged lineages survived in situ with limited gene exchanges, and the stable demographics of lineages indicate that the Pleistocene climatic oscillations were inconsequential for this species. The analysis of genetic variation in populations of R. omeimontis contributes to the understanding of the effects of changes in climate and of geographical events on the dynamic development of contemporary patterns of genetic variation in the species of southern China.
Similar content being viewed by others

Background
Recently, two primary drivers, namely, Pleistocene climatic oscillations and geological events, have been used to explain the contemporary genetic variation within Northern Hemisphere species. In the Pleistocene, climatic cycles generally caused populations to diverge and produce new lineages, additionally affecting the demographics of the populations. In concrete terms, the species were restricted to refugia during glacial periods and subsequently expanded into new habitats during interglacial periods. As a result, the geographic distributions of populations and the genomes of species were markedly affected [1, 2]. However, genetic differentiation is affected differently by geological events [3, 4]. The formation of geographical barriers, including the uplift of range systems, resulted in limited gene flow between populations, thereby providing opportunities for divergence due to genetic drift and natural selection [5, 6]. The current genetic variation within a species is shaped by these two processes working singly or in combination [2, 7, 8].
The indigenous species of southern China are diverse and have complex evolutionary histories. The unique geographic and climatic features in this area most likely shaped the distributions of genetic variation for these species [4, 7, 9]. First, the terrain of southern China is extremely complex and is characterized by numerous mountains and deep valleys such as the Yun-Gui Plateau (YGP) and the Sichuan Basin. The three-stepped geomorphology of the Chinese mainland, bounded by the Kunlun-Qilian-Hengduan mountain ranges and the Daxinganling-Taihang-Wushan-Xuefeng (the western side of the YGP) mountain ranges, is a salient feature of this region [10, 11]. Topographic features can serve as geographic barriers to prevent gene flow between populations and therefore facilitate genetic divergence [4, 12–14]. Second, three to five intermittent glaciations during the Pleistocene have been recorded in the high mountains of southern China [15–17]. Cycles of glaciations can drive postglacial expansion of populations and therefore affect the patterns of genetic variation [9, 18–20]. Thus, species native to southern China are appropriate models to clarify the contributions of geography and climate to contemporary genetic divergence and diversification.
However, for the species in southern China, few studies have focused on the mechanisms that shaped the current patterns of genetic variation, and the relative importance of these mechanisms remains controversial. In studies of birds such as the great tit (Parus major) [20] and the Chinese Hwamei (Leucodioptron canorum canorum) [9], lineage diversification and population expansion during the Pleistocene, reveal the large effects of climatic changes on the genetic variation within these species. However, studies of other species have found that geological events were predominant in affecting genetic variation and that the role of Pleistocene climatic changes was relatively weak. For example, studies on cold water fish and frogs show that genetic lineage divergence coincided with tectonic events (e.g., the uplift of the Ailao Mountains and the QTP), and the lineages were persistent and stable or experienced partial secondary contact during the Pleistocene [4, 7, 8]. These discrepancies in the importance of geography and climate can be attributed to two factors. First, the relative contributions of climate and geology to species genetic variation most likely differ because species inhabit different regions in southern China with diverse topographies and microhabitats. For example, the species that live in continuous mountains responded differently to the glacial oscillations from those that live in isolated mountains, with possibly different population demographics within a species [19]. Second, species with different levels of mobility most likely responded differently to historical processes. For example, highly mobile species (e.g., birds and rats) were more likely to escape from inhospitable habitats to refuges during glaciations and then to disperse long distances to new habitats after ice ages; the result was structured genetic diversity patterns and expanded demographics [19, 21]. In contrast, species with low mobility were likely to remain in situ and consequently were more sensitive than highly mobile species to topographical changes. Thus, further studies are required for a comprehensive understanding of the influences of Pleistocene climate changes and geological events on genetic variation in the species of southern China.
Amphibians are ideal organisms for detecting the effects of various processes on genetic variations [22]. The Omei tree frog (Rhacophorus omeimontis), an arboreal anuran endemic to the subtropical mountain forests of southern China [23], was selected as the object of study because of particular advantages. First, previous research based on fossil records has suggested that Rhacophorid species dispersed from India to Asia at 46 to 57 Ma B.P. [24]. This long-term persistence in Asia allows them to harbor ample genetic legacy of evolutionary processes. Second, unlike the amphibians that spend much of the time in streams or ponds, such as Q. boulengeri (dwells in cold montane streams) [7], adult Omei tree frogs typically live on land. Therefore, these terrestrial frogs are suitable to detect the effects of shifts in the Pleistocene climate because they are more likely affected by environmental changes than aquatic species. Third, their low mobility makes them uneasy to disperse through geographical barriers and thus facilitating genetic divergence [25], which means that they are more likely to be affected by geological events.
In this paper, we determined the contributions of geological events and Pleistocene climatic changes to the contemporary genetic patterns of R. omeimontis with analyses of mitochondrial DNA (mtDNA) sequences (cytochrome b, cytb and NADH dehydrogenase subunit 2, ND2) and nine microsatellite loci. Specifically, if the geological events are the most important driver, then significant genetic differentiation between lineages is expected. In this case, the lineages diverged concurrently with geological events and remained stable during the Pleistocene. Alternatively, the Pleistocene climatic shifts likely contributed to genetic diversification during the evolution of R. omeimontis when the lineage divergence coincided with the Pleistocene glaciations, and/or significant expansion occurred after glacial periods.
Methods
Ethics statement
The animal experiments were performed under an animal ethics approval granted by the School of Life Sciences, Central China Normal University. Our sampling procedures did not significantly affect the survival of the studied species.
Sample collection
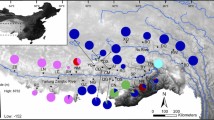
From seven localities in the montane forests of southern China, a total of 196 samples were obtained (Fig. 1). The individuals were all used for mtDNA sequencing, and 175 of them were used for microsatellite allele genotyping (Table 1). For each locality, tissues from at least 20 specimens were collected for DNA analyses. All samples were collected with the permission of the local forestry bureaus or the natural reserves. Specifically, for localities in Sichuan Province, sampling was permitted by the EMeiShan (EMS) city and HongYa (HY) country forestry bureaus, the Fengtongzhai National Natural Reserve in BaoXing (BX) city, and the Laojunshan National Natural Reserve in YiBin (YB) city. Sampling in ZhangJiaJie (ZJJ, Hunan Province) was granted by the Badagongshan National Natural Reserve. Sampling in LongSheng (LS, Guangxi Province) and PingBian (PB, Yunnan Province) were permitted by the Huaping National Natural Reserve and the Daweishan National Natural Reserve, respectively. The sampling localities on the second terrain step of China were EMS, HY, BX and YB of Sichuan Province and PB of Yunnan Province. The populations of ZJJ and LS were located on the third terrain step (Fig. 1). Toe tips were clipped from R. omeimontis (which is evaluated as least concern on the IUCN red list of threatened species, http://www.iucnredlist.org/) to provide the tissue specimens. An antiseptic was used both on the toes and the scissors to reduce infections. The frogs were released immediately after the surgery and the tissue samples were immersed in 95 % ethyl alcohol.

Sampling locations of the populations of R. omeimontis. The locality codes and coordinates are presented in Table 1. The three terrain steps in China and the primary mountain systems are marked. The yellow shadow areas represent the distribution range of R. omeimontis on the IUCN range map (http://www.iucnredlist.org/). The genetic lineages are labeled red, green and blue for the lineages SC, YN and HG, respectively
Laboratory protocols
The total DNA was extracted from the toe tips using a TIANamp Genomic DNA kit (TIANGEN Biotech Co. Ltd., Beijing, China) according to the manufacturer's protocol. We designed two pairs of primers to amplify the fragments of mitochondrial cytb (ROcytb1, CCGCAACACGTAAATCTCAC; ROcytb2, GGGGTGAAGTTATCTGGGTC) and ND2 (ROND2L, GGCCCATACCCCAACAAT; ROND2R, GGCTTTGAAGGCCTTTGG). The PCR was conducted in a 30 μl reaction volume that contained 1 μl of genomic DNA, 15 μl of Premix ExTaq (TaKaRa, Kusatsu, Japan), 1.2 μl of forward and reverse primers and 11.6 μl of ddH2O. All reactions were conducted as follows: an initial denaturation at 94 °C for 3 min, followed by 33 cycles of PCR [94 °C/1 min, 53 °C (cytb) or 55 °C (ND2)/1 min, 72 °C/1 min] and a final extension at 72 °C for 10 min. After a distinct band of the expected fragment size was confirmed, the PCR products were sent to a commercial sequencing company (Nuosai Biotech Inc., Wuhan, China) for sequencing in both directions using an ABI 3730XL DNA Analyzer (Applied Biosystems, Foster, CA, USA). The identical PCR primers were used for the sequencing.
Nine microsatellite loci (OMTF1, OMTF3, OMTF4, OMTF5, OMTF6, OMTF7, OMTF9, OMTF10, and OMTF11) were selected from 11 previously published R. omeimontis loci based on the polymorphic evaluations of each locus [26]. The forward primers were all marked with fluorescent dye (FAM, HEX and TAMRA). The PCR amplifications were conducted in a total volume of 10 μl that contained 0.3 μl of genomic DNA, 5 μl of Premix rTaq (TaKaRa, Kusatsu, Japan), 0.4 μl of forward and reverse primers and 3.9 μl of ddH2O. The reactions began at 95 °C for 5 min, followed by a series of 31 cycles (94 °C/30 s, Tm/30 s, 72 °C/45 s) and a final extension of 72 °C for 10 min. The annealing temperature (Tm) was set within 53–60 °C (for details, see Additional file 1: Table S1). The positive products were assessed using an ABI 3730XL DNA Analyzer (Applied Biosystems, Foster, CA, USA) and scanned using GeneMapper 4.0 (Applied Biosystems, Foster, CA, USA). The results were read with GeneMarker 1.3 (SoftGenetics, State College, PA, USA) and manually input into Microsoft Excel for format transformation and analyses.
Data analyses
Raw data processing
For the mtDNA data, both directions of the cytb and ND2 sequences were assembled into one contig using SeqMan Promode in LaserGene 7.1.0 (Dnastar, Inc.). The sequences were all aligned using ClustalW in MEGA 5.0 [27]. A partition-homogeneity test was conducted in PAUP* 4.10b [28] with 1000 replicates to determine whether the cytb and ND2 fragments could be concatenated into one dataset [29]. Because the results of the test were not significant (p = 0.228), the combined sequences were used in subsequent analyses. For the microsatellite data, the allele values that represented lengths of amplified fragments were transformed into the Arlequin frequency, GenePop2-digit and Fstat formats in Excel using the Microsatellites add-in (MS tools.xla, [30]). Before the analyses, each locus was tested for linkage disequilibrium and deviation from Hardy-Weinberg equilibrium using Genepop (http://genepop.curtin.edu.au/). Significance thresholds were adjusted using the Bonferroni correction. Genotyping errors, including stuttering errors, null alleles and large allele dropouts, were detected using Micro-Checker 2.2.3 [31].
Genetic diversity and population differentiation
Genetic diversity indices were calculated for both the mtDNA sequences and microsatellite data. For the mtDNA sequences, nucleotide diversity (π, [32]) and haplotype diversity (h, [33]) were calculated in DNASP 5.0 [34]. For the microsatellite data, molecular diversity indices, including the number of alleles (N.A.) and the observed (Ho) and expected heterozygosities (He), were calculated in Arlequin 3.1 [35]. Allelic richness (A R) which excludes the influences of sample size was calculated in FSTAT 2.9.3.2 [36].
We then evaluated the genetic variation of populations of R. omeimontis. First, pairwise F-statistics were estimated to evaluate the level of genetic differentiation between population pairs. The Фst (for molecular sequence data) and Rst (for microsatellite data) are useful F-statistics in which they evaluate information not only on the particular haplotypes/alleles frequency but also on the mutational distance [37]. The estimations of pairwise Фst (Jukers & Cantor distance matrix) and Rst (sum of squared differences, Rst-like) values were conducted in Arlequin 3.1 with 10,000 permutations. Then, we analyzed the isolation by distance (IBD) pattern across the seven populations. The Euclidean geographic distances between population pairs were estimated using ArcGis 9.3 [38]. Mantel tests between the geographical distances and the Slatkin linearized Фst (Rst) matrices were conducted in Arlequin 3.1 with 10,000 permutations. Moreover, we conducted a hierarchical analysis of the molecular variance (AMOVA [39]) in Arlequin 3.1 with 10,000 permutations. The population groups were defined based on the results of phylogenetic analyses. Additionally, to intuitively investigate the genetic divergence among genetic lineages, we calculated pairwise genetic distances for the combined and the cytb sequences based on the Kimura 2-parameter model as implemented in MEGA 5.0 (with 10,000 bootstraps).
Matrilineal genealogy and genetic structure
According to the large-scale phylogeny of Amphibia and Rhacophoridae [24, 40], three related species, R. schlegelii, Polypedates megacephalus and Buergeria buergeri (GenBank Accession numbers: AB202078.1, AY458598.1 and AB127977.1, respectively), were selected as the out-groups. These sequences were aligned with our data and combined into an entire dataset. Before reconstruction of the phylogenetic trees, the best-fitting nucleotide substitution model (the general time reversible + proportion of invariable sites + gamma distribution, GTR + I + G) was selected using ModelTest 3.7 [41]. The maximum parsimony (MP) and Bayesian inferences (BI) methods were used to identify the phylogenetic relationships among the mitochondrial haplotypes. The MP tree was constructed in PAUP*4.10b. Heuristic searches were executed for 1000 random addition replicates. The BI tree was constructed in MrBayes 3.1.1 [42]. The Markov chain Monte Carlo (MCMC) analysis was initially performed with four chains at a temperature of 0.2, with the chains run for 5,000,000 cycles. Additionally, the haplotype median-joining network was constructed using the MP method in NETWORK 4.6.1.2 (Fluxus Technology, Suffolk, UK).
The genetic relationships of these populations based on the microsatellite data were deduced using a Bayesian clustering method implemented in STRUCTURE 2.3.1 [43]. The admixture model and the allele frequencies correlated model were used to estimate the number of probable clusters (K value). The lengths of the MCMC iterations were set to 100,000 with a burn-in period of 100,000. During the run, the range of K values was set to 1 to 12, and each K was independently run 20 times. The most probable K value was selected according to the peak value of the average log likelihood [Ln P(X/K)] and the ΔK statistic for a given K [44].
Estimation of divergence time between lineages
To estimate the divergence time between lineages, a suitable molecular clock was established using the relative rate tests in PHYLTST [45]. The best-fitting substitution model was assessed in ModelTest 3.7. A range of substitution rates of 0.65–1.00 % per Ma, which included the mtDNA substitution rates of most amphibians [46–49], was used to calculate the divergence time among lineages in BEAST 1.5.4 [50]. Analyses were conducted for 30 million generations using a uniform speciation (Yule) for each prior tree. Each parameter was evaluated in TRACER 1.5 based on the effective sample size (ESS) value. To ensure the accuracy of the results, we combined three independent results using LogCombiner 1.5.4 and constructed the time tree in TreeAnnotator 1.5.4 using a 10 % burn-in period.
Demographic analyses
We applied three common methods to assess the demographic histories of the three lineages that were inferred from the phylogenetic trees. First, a mismatch distribution was simulated for each lineage to test the hypothesis of recent population growth. A goodness-of-fit test was used to determine the smoothness of the observed mismatch distribution (using Harpending's raggedness index, Rag) and the degree of fit between the observed and simulated data (using the sum of squares deviation, SSD) [51]. Generally, a smooth and unimodal distribution pattern with non-significant p-values for the SSD implies a significant expansion signal for a population. Second, the neutrality tests including Tajima's D [52] and Fu’s Fs [53] were conducted to estimate the demographic expansions of the lineages. In general, significant negative values of Tajima's D and Fu's Fs imply sudden demographic growth. These two analyses were implemented in Arlequin 3.1 with 10,000 bootstrap replicates. Third, Bayesian skyline plots (BSPs) were generated for all combined sequences of each lineage excluding the out-groups in BEAST 1.5.4, which is a method that reliably reconstructs changes in effective population size across evolutionary time scales from sequence data [54]. The suitable substitution models for the three lineages were selected according to Modeltest 3.7. The analyses were conducted with identical settings to those used for the estimation of divergence time, with the exception that the prior tree was set as the coalescent: Bayesian Skyline.
Results
Genetic diversity and population differentiation
For the mtDNA sequencing, a combined mtDNA sequence that included 672 bp cytb and 988 bp ND2 sequences was obtained from each of the 196 individuals. No insertions, deletions or stop codons were found in the entire sequences. Thirty-nine haplotypes were identified and deposited in GenBank (Accession Nos. KP895611-KP895688). For the entire population, the haplotype and nucleotide diversity were relatively high (h = 0.95, π = 1.91 %). Among the seven populations, the haplotype and nucleotide diversity of the BX, LS and PB populations were lower than those in the other four populations (Table 1). For the microsatellite genotyping, artificial errors such as large allele dropouts and stuttering were not detected. The OMTF5 locus presented null alleles and significant deviation from Hardy-Weinberg equilibrium (HWE) across all populations. Of the nine loci, three pairs (OMTF3 & OMTF4, OMTF4 & OMTF5 and OMTF5 & OMTF1) showed significant linkage disequilibrium. Therefore, to guarantee the reliability of results, we excluded two loci (OMTF4 and OMTF5) in the subsequent analyses. For the entire population, the genetic heterozygosity was relatively high (Ho = 0.71, He = 0.87, Table 1). Across the seven populations, the number of microsatellite alleles (N.A.) ranged between 6.71 and 12.14, and the ranges of the Ho, He and A R value were 0.57–0.79, 0.69–0.83 and 6.45–10.37, respectively.
Among the thirty-nine haplotypes identified, only five haplotypes (H3, H4, H5, H7 and H8) were shared by two populations (EMS and HY), with the remaining haplotypes (34 of 39) restricted to one population (see Additional file 2: Table S2). Thus, based on these results, the genetic differentiation among populations was extensive. To further evaluate the genetic differentiation among populations, we conducted pairwise F-statistics. The patterns based on the mtDNA and the microsatellite analyses were similar. The genetic differentiation between all population pairs was significantly different from zero except for the four populations in Sichuan Province (EMS, HY, BX and YB), indicating detectable genetic differentiations (Table 2). Additionally, significant correlations between geographical distances and the Slatkin linearized Фst (Rst) matrices were detected (Table 3), which suggested that the genetic differentiation among populations was correlated with their geographical distributions.
Genetic structure
With similar tree topology from the MP and BI analyses, the seven populations of R. omeimontis were divided into three lineages (the BI tree is shown in Fig. 2a). The haplotypes of the populations ZJJ (Hunan Province) and LS (Guangxi Province), which were distributed in the third terrain step, were clustered into the HG lineage. The haplotypes from the populations in Yunnan and Sichuan provinces in the second terrain step were grouped into a clade with two sub-lineages: one lineage (YN) with the haplotypes of the PB population in Yunnan Province and the other (SC) with the haplotypes from the populations of EMS, HY, BX and YB, which were all in Sichuan Province (Fig. 2a). These three lineages were strongly correlated with the geographic distributions of the populations. The haplotype network also displayed three multiple-mutated clusters, which were similar to those inferred from the phylogenetic trees (Fig. 2b).

Phylogenetic relationships and haplotype network based on the mtDNA haplotypes of R. omeimontis. (a) Phylogenetic tree from Bayesian inference analysis with Bayesian posterior probabilities/maximum parsimony bootstrap values near the branches; and (b) Median-joining network with node sizes proportional to the frequencies of haplotypes. The numbers of mutations separating the haplotypes are shown on the branches, except for the one-step mutations. Empty nodes indicate undetected haplotypes
The genetic clustering analysis based on the microsatellite data produced a genetic structure that was similar to that of the matriline genealogy and haplotypes network of the mtDNA data. As displayed in Fig. 3, the entire population was divided into three genetic clusters (SC, HG and YN) with a distinct maximum ΔK (ΔK = 106.22 at K = 3, Fig. 3b), although a small peak also occurred at K = 6 (ΔK = 29.51) (Fig. 3c). These genetic clusters detected by STRUCTURE analysis were consistent with those of the phylogenetic analyses. To intuitively investigate the genetic divergence among the primary lineages, the pairwise average distances were calculated. For the combined sequences, the genetic distances among the primary lineages varied from 1.4 to 2.2 % (Table 4). Because most studies calculate genetic distances using cytb sequences, we also calculated the genetic distances for cytb sequences. The genetic distances of cytb sequences varied from 0.9 to 2.6 % (Table 4). This range is close to the genetic distances in amphibian con-generic species (2–19 %) [55], reflecting a relatively long-term isolation. Furthermore, from the hierarchical AMOVA, the differentiations among lineages made great contributions to the overall genetic variation of these populations (explained 94.67 % of the total variation in mtDNA and 34.90 % in microsatellite data; Additional file 3: Table S3a, b).

STRUCTURE clustering results deduced from microsatellite alleles within populations of R. omeimontis. (a) Ln P(X/K) and ΔK values as a function of the K values according to 20 run outputs; (b) STRUCTURE results at K = 3, with different colors representing different clusters; and (c) STRUCTURE results at K = 6, with different colors representing different sub-clusters
Estimation of divergence time
Because the hypothesis of clock-like evolution was not rejected at the 5 % level in the relative-rate tests, we set a strict molecular clock model for the analyses. As in the phylogenetic tree analyses, the substitution model was set to GTR + I + G. The time since the most recent common ancestor (tmrca) of the entire in-group of R. omeimontis was estimated at 13.37 Ma [95 % Highest Posterior Density (HPD), 10.14–17.55 Ma; Fig. 4]. The HG lineage was the first to diverge from the other lineages at approximately 2.15 Ma (95 % HPD, 1.56–2.90 Ma), which was followed by the second divergence between the YN and SC lineages at approximately 2.00 Ma (95 % HPD, 1.45–2.71 Ma). The tmrca for B. buergeri and R. omeimontis was estimated to be 42.59 Ma (95 % HPD, 32.63–56.88 Ma), which is close to the previous estimation (45.9 Ma, with 95 % HPD, 36.6–57.6 Ma) [56]; the similarity indicates that our results are reliable.

Estimation of divergence time for R. omeimontis. The numbers near the primary branches are the estimated split times with 95 % highest posterior density. The three primary lineages are indicated by different colors: red, SC lineage; green, YN lineage; and blue, HG lineage
Demographic history
Three methods were used to infer the demographic history of each lineage. For the SC lineage, Tajima's D and Fu's Fs were negative, but not significant (Table 5), and therefore, the rapid expansion hypothesis was rejected. In the mismatch distribution analysis, although a sudden expansion was not rejected by the goodness-of-fit tests based on non-significant SSD and Rag values, the sudden expansion hypothesis was not supported because the frequency distribution expressed a multimodal pattern with an initial peak followed by the maximum height and a subsequent small peak (Fig. 5a). Based on the Bayesian skyline plots (BSPs), the effective population size remained stable during the interglacial period (LIG, 0.075–0.125 Ma) [16] and then decreased slightly at approximately 0.012 Ma (early stage of the Holocene) (Fig. 5d). Therefore, signals for significant expansion were not detected in the SC lineage. Similarly, based on the neutrality tests and mismatch distribution analyses (Table 5 and Fig. 5b, c), the rapid expansion hypothesis was rejected for the HG and YN lineages. Additionally, the effective population sizes of these two lineages remained stable over evolutionary timescales as indicated by the BSP curves (Fig. 5e, f).

Demographic history analyses for the three lineages according to mismatch distributions and Bayesian skyline plots. (a, b and c) Mismatch distributions for the SC, HG and YN lineages, respectively. The x coordinate represents the number of differences in each pair of sequence comparisons, and the y coordinate represents the frequencies of pairwise differences. (d, e and f) Bayesian skyline plots for the SC, HG and YN lineages, respectively. The x-axis indicates time in Ma BP, and the y-axis indicates the effective population size in units of Neτ (the product of effective population size and generation time in Ma). The blue areas represent 95 % highest posterior density
Discussion
The Omei tree frog (R. omeimontis) is restricted to subtropical forests in southwest China [23]. In this study, based on specimens collected in seven primary areas within the distribution of R. omeimontis, we found that the entire population was highly genetically diverse. The high level of mtDNA haplotype diversity for R. omeimontis (h = 0.95, π = 1.91 %) is comparable with that of other amphibians in China, such as Quasipaa boulengeri (h = 0.96, π = 0.97 %) [7] and Buergeria robusta (h = 0.98, π = 1.14 %) [57]. However, the genetic diversity based on microsatellite data was higher in R. omeimontis (Ho and He equal to 0.71 and 0.87, respectively) than that in other amphibians such as the moor frog (Rana arvalis, Ho = 0.34 and He = 0.38) [58] and the wood frog (Rana sylvatica, Ho = 0.60 and He = 0.64 for the highest polymorphic locus) [59]. These high levels of overall genetic diversity were a reflection of the deep divergence among populations.
Pairwise F-statistics were calculated to evaluate the genetic differentiation between populations based on the mtDNA sequences and the microsatellite markers. A significant deviation from zero was detected for both Фst and Rst in all population pairs except for the pairwise Rst between populations obtained from Sichuan Province, which indicated detectable genetic differentiation between R. omeimontis populations. The incongruence of differentiation pattern between the mtDNA and microsatellite data could be attributed to the different types of inheritance and rates of evolution for the two types of markers and to the unique dispersal pattern of R. omeimontis. The polyandry mating system of R. omeimontis and the observation that the return rate of R. omeimontis females is much higher than males [25, 60] suggest intense female philopatry and historical male dispersal for breeding. Therefore, scarce female gene flow could result in significant mtDNA (maternal inheritance) genetic differentiation among populations, whereas the relatively frequent male dispersal could diminish the genetic differences in microsatellites (parental inheritance). This male-biased gene flow has been reported in many other vertebrates, including birds, lizards and frogs [13].
Based on the broadly consistent results inferred from the mtDNA markers and the microsatellite loci, the seven populations of R. omeimontis were divided into three genetic lineages (SC, YN and HG) (Fig. 2) with deep divergences. This genetic structure recovered within R. omeimontis was consistent with the complex geological systems in southern China (Fig. 1). Specifically, the SC and YN lineages (located in Sichuan and Yunnan provinces, respectively) and the HG lineage (distributed across Hunan and Guangxi provinces) were separated in the second and third terrain steps of southern China, respectively. Additionally, the SC lineage was restricted to the Sichuan Basin, and the YN lineage was restricted to the YGP (Fig. 1). Moreover, the validity of this geographically structured pattern was supported by the large mutation steps among the haplotype networks (Fig. 2b) and the relatively high genetic distances among the three lineages (Table 4). These results suggest that the genetic lineages diverged deeply and that the divergence might be largely related to specific geologic events within this region. The first splitting event between the HG lineage and the ancestor of the SC and YN lineages was dated to approximately 2.15 Ma with a 95 % HPD of 1.56–2.90 Ma. This splitting event occurred during the periods of recent intense movements of the QTP (1.7–3.6 Ma), which are largely responsible for the formation of the three-stepped terrain in China [11, 61]. The formation of the three-stepped terrain most likely generated geographic barriers that prevented gene flow between the HG lineage and the other populations. Subsequently, dragged by the uplift of the QTP, the YGP also elevated and experienced strong tectonic movements and frequent dry-humid oscillations at 1.4–2.5 Ma [62]. This period of changes for the YGP was consistent with the second splitting event between the SC and YN lineages that occurred at approximately 2.00 Ma (with 95 % HPD, 1.45–2.71 Ma). The tectonic and climatic changes of the YGP might have isolated the YN lineage from the SC lineage and led to the accumulation of autapomorphic mutations. Therefore, the two splitting events within R. omeimontis were most likely associated with successive tectonic events (including the recent uplift of the QTP and the subsequent movements of the YGP). Each phase of these movements possibly generated fragmented habitats, which could facilitate local divergence because of limited gene flow among populations for a species with such a low mobility. Similar patterns are also reported in Leiolepos reevesii [63], Leptobrachium ailaonicum [8], Rhynchocypris oxycephalus [4] and Babina pleuraden [64]. Hence, the complex geological events were most likely an important factor that drove the lineage divergence of species in southern China.
However, notably, the splitting events within R. omeimontis were estimated to have occurred during the Pleistocene (extended from 2.59 Ma to 0.012 Ma [65]) which characterized by cyclic glaciations [1, 4]. The repeated spells of cooling in the Pleistocene could also have increased divergence of the genetic lineages in this environmentally sensitive species. Faced with harsh climates, the species was most likely restricted to several suitable habitats. Combined with the influences of the topographic barriers shaped by geographic events, these populations of R. omeimontis were more prone to differentiate rapidly and exhibit deep genetic divergence among lineages; thus, an alternative explanation is provided for the two recent splitting events and the relatively large genetic distances among lineages. Therefore, coupled with the intense tectonic movements, the Pleistocene climatic oscillations were most likely another important factor driving the divergence of the lineages of R. omeimontis.
After diverging from one another, the lineages of R. omeimontis apparently survived in situ to shape the current geographically structured genetic pattern. The gene flow was most likely limited among lineages based on the low genetic similarity and the deep genetic differentiation revealed by the genetic structure analyses. When combined with the low migration capability of this species, the presumption is geographic barriers hindered gene flow and thereby resulted in large genetic differentiation and local evolution. Furthermore, each lineage remained stable during the local evolution. As shown by the demographic history reconstruction, the effective population sizes of all lineages remained constant through evolutionary time. Both the mismatch analyses and neutrality tests did not detect significant signals of rapid expansion, which are the typical responses to climatic oscillations [9, 18–20]. Thus, the Pleistocene climatic oscillations might have had no detectable influences on the demographics of this species. Similarly, for the spiny frog, Quasipaa boulengeri, the long-term demographics of lineages during the Pleistocene were also stable in the southern China [7]. Additionally, with distributions in regions identical to those considered in this study, some bird populations remained stable during the Pleistocene, despite population expansion in other regions [19, 66]. Thus, the less significant effects of Pleistocene climatic cycling on demographics might be common to the species in southern China. This phenomenon might be attributed to the buffering effects of topographic features in southern China because the complex mosaic of mountains and valleys provided numerous habitable microhabitats during the Pleistocene ice ages for species to escape from cold climate conditions in situ in southern China [9, 67, 68].
Conclusions
In this paper, we investigated the genetic structure of the populations of R. omeimontis and evaluated the relative roles of Pleistocene climatic oscillations and geological events in shaping the current patterns of genetic differentiation of the species in southern China. The levels of genetic diversity were high, and the populations of R. omeimontis were clearly differentiated. The phylogenetic reconstructions and the estimation of divergence time indicated that complex geological events (including the recent uplift of the QTP and the subsequent movements of the YGP) coupled with the Pleistocene glaciations drove the genetic divergence of the lineages of R. omeimontis. Following the divergence, each lineage was restricted in situ and diverged independently without distinct population expansion. Therefore, the Pleistocene climatic oscillations did not appear to influence the demographics of this species, and this phenomenon might be unique for species in general in southern China because of the complex topography. The results of this study will increase our comprehensive understanding of the relative contributions of geographical events and the Pleistocene climatic changes to the current genetic patterns of species in southern China.
Availability of supporting data
The sequence dataset generated herein is available in the GenBank repository with Accession numbers KP895611-KP895688.
References
Hewitt GM. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–13.
Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Phil Trans R Soc Lond B. 2004;359:183–95.
Wu YK, Wang YZ, Jiang K, Hanken J. Significance of pre-Quaternary climate change for montane species diversity: insights from Asian salamanders (Salamandridae: Pachytriton). Mol Phylogenet Evol. 2013;66:380–90.
Yu D, Chen M, Tang QY, Li XJ, Liu HZ. Geological events and Pliocene climate fluctuations explain the phylogeographical pattern of the cold water fish Rhynchocypris oxycephalus (Cypriniformes: Cyprinidae) in China. BMC Evol Biol. 2014;14:225.
Che J, Zhou WW, Hu JS, Yan F, Papenfuss TJ, Wake DB, et al. Spiny frogs (Paini) illuminate the history of the Himalayan region and Southeast Asia. Proc Natl Acad Sci USA. 2010;107:13765–70.
Kieswetter CM, Schneider CJ. Phylogeography in the northern Andes: Complex history and cryptic diversity in a cloud forest frog, Pristimantis w-nigrum (Craugastoridae). Mol Phylogenet Evol. 2013;69:417–29.
Yan F, Zhou WW, Zhao HT, Yuan ZY, Wang YY, Jiang K, et al. Geological events play a larger role than Pleistocene climatic fluctuations in driving the genetic structure of Quasipaa boulengeri (Anura: Dicroglossidae). Mol Ecol. 2013;22:1120–33.
Zhang MW, Rao DQ, Yang JX, Yu GH, Wilkinson JA. Molecular phylogeography and population structure of a mid-elevation montane frog Leptobrachium ailaonicum in a fragmented habitat of Southwest China. Mol Phylogenet Evol. 2010;54:47–58.
Li SH, Yeung CKL, Feinstein J, Han LX, Le MH, Wang CX, et al. Sailing through the Late Pleistocene: unusual historical demography of an East Asian endemic, the Chinese Hwamei (Leucodioptron canorum canorum), during the last glacial period. Mol Ecol. 2009;18:622–33.
Jiang F, Wu X. Fundamental characteristics of the stepped landform in China continent (In Chinese). Mar Geol & Quat Ge. 1993;13:15–24.
Zhang DF, Fengquan L, Jianmin B. Eco-environmental effects of the Qinghai-Tibet Plateau uplift during the Quaternary in China. Environ Geol. 2000;39:1352–8.
Gao LM, Möller M, Zhang XM, Hollingsworth ML, Liu J, Mill RR, et al. High variation and strong phylogeographic pattern among cpDNA haplotypes in Taxus wallichiana (Taxaceae) in China and North Vietnam. Mol Ecol. 2007;16:4684–98.
Yan J, Wang QX, Chang Q, Ji X, Zhou K. The divergence of two independent lineages of an endemic Chinese gecko, Gekko swinhonis, launched by the Qinling orogenic belt. Mol Ecol. 2010;19:2490–500.
Zhao Q, Liu HX, Luo LG, Ji X. Comparative population genetics and phylogeography of two lacertid lizards (Eremias argus and E. brenchleyi) from China. Mol Phylogenet Evol. 2011;58:478–91.
Zheng B, Xu Q, Shen Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai-Tibetan Plateau: review and speculation. Quatern Int. 2002;97:93–101.
Zhao J, Shi Y, Wang J, Du J, Yang Z, Shi L. 867 Comparison between Quaternary Glaciations in China and the Marine Oxygen Isotope Stage (MIS): An Improved Schema (In Chinese). 2011.
Li JJ, Shu Q, Zhou SZ, Zhao ZJ, Zhang JM. Review and prospects of Quaternary glaciation research in China (In Chinese). J Glaciol Geocryol. 2004;26:235–43.
Brito PH. The influence of Pleistocene glacial refugia on tawny owl genetic diversity and phylogeography in western Europe. Mol Ecol. 2005;14:3077–94.
Song G, Qu YH, Yin ZH, Li S, Liu NF, Lei FM. Phylogeography of the Alcippe morrisonia (Aves: Timaliidae): long population history beyond late Pleistocene glaciations. BMC Evol Biol. 2009;9:143.
Zhao N, Dai C, Wang W, Zhang R, Qu Y, Song G, et al. Pleistocene climate changes shaped the divergence and demography of Asian populations of the great tit Parus major: evidence from phylogeographic analysis and ecological niche models. J Avain Biol. 2012;43:297–310.
Chen W, Liu S, Liu Y, Hao H, Zeng B, Chen S, et al. Phylogeography of the large white-bellied rat Niviventer excelsior suggests the influence of Pleistocene Glaciations in the Hengduan Mountains. Zool Sci. 2010;27:487–93.
Beebee TJC. Conservation genetics of amphibians. Heredity. 2005;95:423–7.
Liao WB, Lu X. Breeding behaviour of the Omei tree frog Rhacophorus omeimontis (Anura: Rachophoridae) in a subtropical montane region. J Nat Hist. 2010;44:2929–40.
Li JT, Li Y, Klaus S, Rao DQ, Hillis DM, Zhan YP. Diversification of rhacophorid frogs provides evidence for accelerated faunal exchange between India and Eurasia during the Oligocene. Proc Natl Acad Sci USA. 2013;110:3441–6.
Yang YJ, Lin YS, Wu JL, Hui CF. Variation in mitochondrial DNA and population structure of the Taipei treefrog Rhacophorus taipeianus in Taiwan. Mol Ecol. 1994;3:219–28.
Zhao M, Zhang RP, Li CL, Mu TY, Wei SC, Li X, et al. Development of novel microsatellite markers in the OmeiTreefrog (Rhacophorus omeimontis). Int J Mol Sci. 2012;13:552–57.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods) v. 4.0 b10. Sunderland: Sinauer Associates; 2002.
Farris JS, Källersjö M, Kluge AG, Bult C. Constructing a significance test for incongruence. Syst Biol. 1995;44:570–2.
Park SDE. The excel microsatellite toolkit: Trypanotolerance in West African cattle and the population genetic effects of selection (Ph.D. thesis). Dublin: University of Dublin; 2001.
Van Oosterhout C, Hutchinson WF, Wills PM, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4:535–8.
Nei M, Tajima F. DNA polymorphism detectable by restriction endonucleases. Genetics. 1981;97:145–63.
Nei M. Molecular Evolutionary Genetics. New York: Columbia University Press; 1987.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50.
Goudet J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). [Available from http://www2.unil.ch/popgen/softwares/fstat.htm] The current version is 2.9.3.2 (Feb. 2002).
Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet. 2009;10:639–50.
Environmental Systems Research Institute. ArcGis V.9.3. Redlands, CA: ESRI; 2009.
Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91.
Pyron RA, Wiens JJ. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol Phylogenet Evol. 2011;61:543–83.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8.
Ronquist F, Huelsenbeck J. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Pritchard JK, Stephens M, Donnelly PJ. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Kumar S. Phyltest: A program for testing phylogenetic hypothesis, Version 2.0. Pennsylvania, USA: The Pennsylvania State University; 1996.
Macey JR, Schulte II JA, Larson A, Fang ZL, Wang YZ, Tunniyev BS, et al. Phylogenetic relationships of toads in the Bufo bufo species group from the eastern escarpment of the Tibetan Plateau: a case of vicariance and dispersal. Mol Phylogenet Evol. 1998;9:80–7.
Hoffman EA, Blouin MS. Evolutionary history of the northern leopard frog: reconstruction of phylogeny, phylogeography, and historical changes in population demography from mitochondrial DNA. Evolution. 2004;58:145–59.
Carnaval AC, Bates JM. Amphibian DNA shows marked genetic structure and tracks Pleistocene climate change in northeastern Brazil. Evolution. 2007;61:2942–57.
Lu B, Zheng YC, Murphy RW, Zeng XM. Coalescence patterns of endemic Tibetan species of stream salamanders (Hynobiidae: Batrachuperus). Mol Ecol. 2012;21:3308–24.
Drummond AJ, Rambaut A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Harpending HC. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol. 1994;66(4):591–600.
Tajima F. Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu YX. Statistical test of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22:1185–92.
Johns GC, Avise JC. A comparative summary of genetic distances in the vertebrates from the mitochondrial cytochrome b gene. Mol Biol Evol. 1998;15:1481–90.
Roelants K, Gower D, Wilkinson M, Loader SP, Biju SD, Guillaume K, et al. Global patterns of diversification in the history of modern amphibians. Proc Natl Acad Sci USA. 2007;104:887–92.
Lin HD, Chen YR, Lin SM. Strict consistency between genetic and topographic landscapes of the brown tree frog (Buergeria robusta) in Taiwan. Mol Phylogenet Evol. 2012;62:251–62.
Vos CC, Antonisse-De Jong AG, Goedhart PW, Smulders MJM. Genetic similarity as a measure for connectivity between fragmented populations of the moor frog (Rana arvalis). Heredity. 2001;86:598–608.
Newman RA, Squire T. Microsatellite variation and fine-scale population structure in the wood frog (Rana sylvatica). Mol Ecol. 2001;10:1087–100.
Zhao M, Li C, Zhang W, Wang H, Luo Z, Gu Q, et al. Male pursuit of higher reproductive success drives female polyandry in the Omei treefrog. Anim Behav. 2016;111:101–10.
Li JJ. Studies on the geomorphological evolution of the Qinghai-Xizang (Tibetan) Plateau and Asian monsoon (In Chinese). Mar Geol Quat Ge. 1999;19:1–11.
Tong GB, Zhang JP, Yang XD, Luo BX, Wang YZ, Chen PY. Late Cenozoic palynoflora and environment changes in Yunnan-Guizhou Plateau (In Chinese). Mar Geol Quat Ge. 1994;14:91–104.
Lin LH, Ji X, Diong CH, Du Y, Lin CX. Phylogeography and population structure of the Reevese’s Butterfly Lizard (Leiolepis reevesii) inferred from mitochondrial DNA sequences. Mol Phylogenet Evol. 2010;56:601–7.
Li ZJ, Yu GH, Rao DQ, Yang JX. Phylogeography and demographic history of Babina pleuraden (Anura, Ranidae) in southwestern China. PloS One. 2012;7:e34013.
Gibbard PL, Head MJ, Walker MJ. Formal ratification of the Quaternary System/Period and the Pleistocene Series/Epoch with a base at 2.58 Ma. J Quaternary Sci. 2010;25:96–102.
Qu Y, Ericson PGP, Quan Q, Song G, Zhang R, Gao B, et al. Long-term isolation and stability explain high genetic diversity in the Eastern Himalaya. Mol Ecol. 2014;23:705–20.
Huang Z, Liu N, Liang W, Zhang Y, Liao X, Ruan L, et al. Phylogeography of Chinese bamboo partridge, Bambusicola thoracica thoracica (Aves: Galliformes) in south China: Inference from mitochondrial DNA control-region sequences. Mol Phylogenet Evol. 2010;56:273–80.
Qian H, Ricklefs RE. Large-scale processes and the Asian bias in species diversity of temperate plants. Nature. 2000;407:180–2.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 31201713, No. 31270425, No. 31470442) and the research funds from Central China Normal University (No. CCNU14B01001).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HW conceived of the study, and participated in its design and coordination and helped to draft the manuscript. JL and MZ carried out the molecular experiments, aligned the sequences, analyzed the data and drafted the manuscript. SCW participated in samples collection and doing laboratory experiments. ZHL participated in the design of the study, drew the figures and critically revised the manuscript. All authors read and approved the final manuscript.
Jun Li and Mian Zhao contributed equally to this work.
Additional files
Additional file 1: Table S1.
Types of labeled fluorescent dye, annealing temperature (Tm) and GenBank accession numbers of microsatellite loci. (DOC 32 kb)
Additional file 2: Table S2.
Distribution of combined mtDNA sequence haplotypes in seven populations. (DOC 71 kb)
Additional file 3: Table S3.
a. Hierarchical AMOVA analysis based on mtDNA sequences. b. Hierarchical AMOVA analysis based on microsatellite data. (DOC 33 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, J., Zhao, M., Wei, S. et al. Geologic events coupled with Pleistocene climatic oscillations drove genetic variation of Omei treefrog (Rhacophorus omeimontis) in southern China. BMC Evol Biol 15, 289 (2015). https://doi.org/10.1186/s12862-015-0572-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-015-0572-1