
Abstract
Background
High-throughput amplicon sequencing of environmental DNA (eDNA metabarcoding) has become a routine tool for biodiversity survey and ecological studies. By including sample-specific tags in the primers prior PCR amplification, it is possible to multiplex hundreds of samples in a single sequencing run. The analysis of millions of sequences spread into hundreds to thousands of samples prompts for efficient, automated yet flexible analysis pipelines. Various algorithms and software have been developed to perform one or multiple processing steps, such as paired-end reads assembly, chimera filtering, Operational Taxonomic Unit (OTU) clustering and taxonomic assignment. Some of these software are now well established and widely used by scientists as part of their workflow. Wrappers that are capable to process metabarcoding data from raw sequencing data to annotated OTU-to-sample matrix were also developed to facilitate the analysis for non-specialist users. Yet, most of them require basic bioinformatic or command-line knowledge, which can limit the accessibility to such integrative toolkits. Furthermore, for flexibility reasons, these tools have adopted a step-by-step approach, which can prevent an easy automation of the workflow, and hence hamper the analysis reproducibility.
Results
We introduce SLIM, an open-source web application that simplifies the creation and execution of metabarcoding data processing pipelines through an intuitive Graphic User Interface (GUI). The GUI interact with well-established software and their associated parameters, so that the processing steps are performed seamlessly from the raw sequencing data to an annotated OTU-to-sample matrix. Thanks to a module-centered organization, SLIM can be used for a wide range of metabarcoding cases, and can also be extended by developers for custom needs or for the integration of new software. The pipeline configuration (i.e. the modules chaining and all their parameters) is stored in a file that can be used for reproducing the same analysis.
Conclusion
This web application has been designed to be user-friendly for non-specialists yet flexible with advanced settings and extensibility for advanced users and bioinformaticians. The source code along with full documentation is available on the GitHub repository (https://github.com/yoann-dufresne/SLIM) and a demonstration server is accessible through the application website (https://trtcrd.github.io/SLIM/).
Similar content being viewed by others

Background
High-throughput amplicon sequencing of environmental DNA (eDNA metabarcoding) is a fast and affordable molecular approach to monitor biodiversity [1]. Metabarcoding has indeed become a routinely used tool for various ecological field, such as terrestrial and marine biodiversity studies [2], animals diet survey [3] or biomonitoring [4,5,6]. It has even proved useful for paleo-environmental events detection [7], archeological studies [8], and the detection of airborne pollen [9]. The data generated by sequencing platforms during these studies is being processed by a succession of software (a so-called pipeline) to translate the raw sequences (or reads) into a statistically exploitable contingent matrix that contains Operational Taxonomic Units (OTU) as rows and samples as columns (i.e. the so-called “OTU-table”). These processing steps are indeed critical for accurate biological interpretation [10,11,12,13].
The metabarcoding processing steps can be broadly grouped in five categories:
-
1.
Demultiplexing samples: Most of the metabarcoding studies uses multiplexing for a better cost-effectiveness, i.e. including sample-specific tags in the primers prior PCR amplification [14]. From a given multiplexed library (pooled PCR products with unique adaptors pairs at both 3′ and 5′ ends of the reads), multiple samples need to be retrieved and “demultiplexed” into separate sample-specific files. In the case of each library represents a unique sample, this step can be ignored.
-
2.
Reads joining: For paired-end sequencing data, reads need to be joined into full-length contigs. This step can also be seen as quality filtering, because non-overlapping reads are being discarded. For single-end sequenced libraries, this step can be ignored.
-
3.
Quality filtering: It regroups multiple type of filters, including base-calling quality filters, PCR and sequencing errors denoiser [15] or chimera filter [16]. This step is crucial to remove as much technical noise as possible in the data.
-
4.
OTU clustering: This step has received most of the attention and is still an active field of bioinformatic research. The sequences are grouped by similarity into clusters that represent proxies for molecular species (de novo OTU clustering strategy). Open or closed reference OTU clustering strategies sequences are mapped represent alternatives (sequences are first clustered against a reference sequence database), even though they have been shown to be outperformed by de novo approaches in some cases [17]. This step is critical to yield a maximum of biologically relevant information and has a strong impact on diversity measures and downstream analysis.
-
5.
Taxonomic assignment: Putatively ascribe a taxonomic name to each OTU. Curated sequence databases such as SILVA [18] or PR2 [19] for nuclear ribosomal markers, BOLD [20] or MIDORI [21] for cytochrome oxidase I or UNITE for fungal Internal Transcribed Spacer [22] can be used as reference. Important efforts are made to improve methods and algorithms for more accurate taxonomic assignment, and various approaches have been explored [23,24,25,26].
Multiple algorithms and software have been developed to perform one or multiple processing steps. They can be called sequentially via command-line or bash scripts to form an analysis pipeline, provided that the input/output file format between each of these software is handled correctly. Wrappers and toolkits such as MOTHUR [27], USEARCH [28], QIIME [29], OBITools [30] or VSEARCH [31] have been developed specifically for routine analysis of eDNA metabarcoding data. However, non-specialists or command-line reluctant users might still not feel comfortable. Moreover, users are often left to find by themselves a relevant traceability system for their analysis, which can hamper the analyses reproducibility. The software galaxy [32] was developed to allow users to create their own pipelines through a web Graphical User Interface (GUI). However, it has been designed to remain as broad as possible in term of application. This means going through a long configuration and installation step prior any data analysis. A command-line free tool specifically designed for metabarcoding studies, yet flexible and powerful, would allow every scientist working with such sequencing data to be autonomous for the carry-out of these critical processing steps.
Here, we introduce SLIM, an open-source web application for the reproducible processing of metabarcoding data, from the raw sequences to an annotated OTU table. The application is meant to be deployed on a local computing server or on personal computers for users without internet connection or developers. We provide a demonstration version of SLIM with reduced computing capacity, accessible through the application website (https://trtcrd.github.io/SLIM/).
Implementation
Overview
SLIM is an web-application with a Graphical User Interface (GUI) that help users to create and execute their own metabarcoding pipelines using state-of-art, open-source and well-established software. The core of SLIM is based on the Node JavaScript runtime, an open source server framework that have been designed for the building of scalable network applications, by handling asynchronous and parallel events. The installation is made as easy as possible for system administrators, through bash scripts that fetch the dependencies, and deploy the web application into a docker container (www.docker.com). This means that the application can be deployed on various platform, from a personal computer to a local or cloud-based computing server. The development of SLIM was guided by the four following principles:
Making it user-friendly for non-specialists
This involves creating a Graphical User Interface (GUI) to avoid the need of any command line. For Operating System (OS) cross compatibilities, portability and maintenance, we used web technologies (JavaScript, HTML and CSS) to build the GUI. Therefore, there is no need for any installation on user’s personal computers. Instead, SLIM is accessible through a web-browser over local network or over the internet, from any operating system (OS).
Making the installation and administration as easy as possible
To facilitate the installation and the deployment of SLIM by systems administrators while ensuring the security and stability of a computing server configuration, we embedded the application in a docker container (https://www.docker.com). Thanks to this solution, SLIM can be deployed on Unix-like OS (macOS and Linux). We created two bash scripts, one to fetch the application dependencies and another one to deploy it. The application is versioned and frozen into stable releases hosted in GitHub. Once deployed, SLIM includes a logging system that is accessible through docker commands.
Encouraging analysis reproducibility
Analysis reproducibility and transparency is a growingly recognized issue. We included an easy way to reproduce an analysis carried out by SLIM. Each execution, which includes a succession of software with their associated parameters can be saved and stored as a small configuration file. To exactly reproduce an execution, one just need the raw sequencing data, the stable version of SLIM that has been used and this configuration file.
Facilitating its extensibility
The integration of new software into modules has been made as easy as possible. It requires only some knowledge of web-based languages (JavaScript and HTML) and for input/output file format handling (usually done by python scripts). Once the set of module’s associated files are in place within the application folders, the integration itself is done automatically by the application core functions. Developers are encouraged to pull request their new modules and new features to the SLIM repository (https://github.com/yoann-dufresne/SLIM). These new features will be merged to SLIM and made available on the demonstration server.
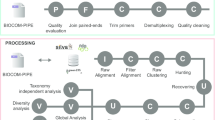
A module-centered application
All the implemented software and tools are independently encapsulated in modules. Each module is defined with its input files, its parameters and its output files. This organization structure makes it possible to create single or parallel workflows to study the impact of a specific step on the biological conclusions, by connecting outputs of modules to inputs of others (Fig. 1). This chaining organization makes SLIM flexible and adapted for a wide range of use cases. Indeed, adding and chaining modules is an intuitive way to design workflows. The processing modules that are readily available in SLIM and the ones that are planned to be included in future development is listed in Table 1. These future modules include for instance a mistagging filter [33], the DADA2 [15] workflow for Amplicon Sequence Variant (ASV) inference, the CREST [24] and IDTAXA [34] taxonomic assignment method, the Short Read Archive (SRA) toolkit for fetching raw data directly from the application, but also some post-processing tools. For instance, the R package LULU that implement a post clustering curation algorithm [35] has been integrated, and the R package BBI for computing Biotic Indices from the taxonomic assignments [36] will be soon available. A complete documentation for each module specifications is available on the SLIM GitHub repository wiki. We also provide a detailed documentation for the development of new modules.

Two pipeline examples using SLIM. A) A commonly used workflow including usual processing steps, from the demultiplexing to an annotated OTU table. B) An alternate workflow using different OTU clustering strategies to assess the impact of this processing step on the biological conclusions
The job execution, data management and queuing system
Once the data is uploaded and the pipeline has been set, users provide their email address and trigger the execution. An email containing a unique link to the job as well as the configuration file will be immediately sent to the user. The job will be automatically scheduled and run. As soon as the job is done, a second email will be sent, inviting the user to download the annotated OTU table and any intermediate file of interest. By default, the raw data and results file will remain available on the server for a period of 24 h after job completion for storage optimization.
The application has been designed to be multi-tenant and to adapt the number of parallel users (i.e. tenant) that can perform an execution to the computing capacity of the hosting machine. By default, we have set the application to execute a user’ job on up to 8 CPU cores (16 cores make it possible to execute two users’ job in parallel, etc.). If a new job is submitted while all the CPU cores are already busy, it will be queued. Queued jobs will be scheduled as soon as enough CPU cores become available.
Results and discussion
SLIM is a user-friendly web application specifically designed for the processing of raw metabarcoding data to obtain annotated OTU tables. It simplifies the use of state-of-art bioinformatics tools, by providing an intuitive GUI that allows users to quickly design their own analysis pipeline. It also facilitates the reproducibility of a such analysis, by sending to the user an email containing a configuration file that includes all the pipeline settings. Hence, reproducing an analysis requires only the raw sequencing dataset, the version of SLIM that was used, and this configuration file. We think that including such configuration file as supplementary material in publications will contribute to improve the reproducibility of metabarcoding analysis.
Thanks to the use of web technologies, SLIM is cross-platform and is meant to be deployed on computing server and accessed remotely over local network or over the internet. However, users with limited internet connection and developers can also install the application on their own personal computer running Unix-like OS (Linux or macOS).
The future development and integration of new modules has been made as easy as possible and will make SLIM even more flexible and useful to the metabarcoding users community. This aspect is of prime importance as sequencing technologies are constantly being improved and keep in challenging our computing tools to extract biologically relevant information from this ever-growing amount of data.
Conclusion
For demonstration purpose, a server is accessible from the project website hosted on GitHub (https://trtcrd.github.io/SLIM/) and has been restricted to process up to one single full illumina MiSeq platform run (approximately 15 million reads) or to execute quickly an analysis on a provided example dataset.
Availability and requirements
Project name: SLIM
Project home page: https://github.com/yoann-dufresne/SLIM
Project demonstration page: https://trtcrd.github.io/SLIM/
Operating system(s): Linux, macOS
Programming language: JavaScript, Python, HTML, CSS, Shell
Other requirements: docker
License: AGPL v3
Abbreviations
- CPU:
-
Computing processing unit
- GUI:
-
Graphical user interface
- OTU:
-
Operational taxonomic unit
- PCR:
-
Polymerase chain reaction
References
Taberlet P, Coissac E, Pompanon F, Brochmann C, Willerslev E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol Ecol. 2012;21:2045–50.
Valentini A, Taberlet P, Miaud C, Civade R, Herder J, Thomsen PF, et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol Ecol. 2016;25:929–42.
Pompanon F, Deagle BE, Symondson WOC, Brown DS, Jarman SN, Taberlet P. Who is eating what: diet assessment using next generation sequencing. Mol Ecol. 2012;21:1931–50.
Lanzén A, Lekang K, Jonassen I, Thompson EM, Troedsson C. High-throughput metabarcoding of eukaryotic diversity for environmental monitoring of offshore oil-drilling activities. Mol Ecol. 2016;25:4392–406.
Apothéloz-Perret-Gentil L, Cordonier A, Straub F, Iseli J, Esling P, Pawlowski J. Taxonomy-free molecular diatom index for high-throughput eDNA biomonitoring. Mol Ecol Resour. 2017;17:1231–42.
Cordier T, Forster D, Dufresne Y, Martins CI, Stoeck T, Pawlowski J. Supervised machine learning outperforms taxonomy-based environmental DNA metabarcoding applied to biomonitoring. Mol Ecol Resour. 2018. https://doi.org/10.1111/1755-0998.12926.
Szczuciński W, Pawłowska J, Lejzerowicz F, Nishimura Y, Kokociński M, Majewski W, et al. Ancient sedimentary DNA reveals past tsunami deposits. Mar Geol. 2016;381:29–33.
Grealy A, Douglass K, Haile J, Bruwer C, Gough C, Bunce M. Tropical ancient DNA from bulk archaeological fish bone reveals the subsistence practices of a historic coastal community in Southwest Madagascar. J Archaeol Sci. 2016;75:82–8.
Leontidou C, Vernesi C, de Groeve J, Cristofolini F, Vokou D, Cristofori A. Taxonomic identification of airborne pollen from complex environmental samples by DNA metabarcoding: a methodological study for optimizing protocols. bioRxiv. 2017:099481. https://doi.org/10.1101/099481.
Lekberg Y, Gibbons SM, Rosendahl S. Will different OTU delineation methods change interpretation of arbuscular mycorrhizal fungal community patterns? New Phytol. 2014;202:1101–4.
He Y, Caporaso JG, Jiang X-T, Sheng H-F, Huse SM, Rideout JR, et al. Stability of operational taxonomic units: an important but neglected property for analyzing microbial diversity. Microbiome. 2015;3:20. https://doi.org/10.1186/s40168-015-0081-x.
Schmidt TSB, Matias Rodrigues JF, von Mering C. Limits to robustness and reproducibility in the demarcation of operational taxonomic units. Environ Microbiol. 2015;17:1689–706.
Forster D, Dunthorn M, Stoeck T, Mahé F. Comparison of three clustering approaches for detecting novel environmental microbial diversity. PeerJ. 2016;4:e1692. https://doi.org/10.7717/peerj.1692.
Binladen J, Gilbert MTP, Bollback JP, Panitz F, Bendixen C, Nielsen R, et al. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One. 2007;2:e197.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. https://doi.org/10.1038/nmeth.3869.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200.
Westcott SL, Schloss PD. De novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ. 2015;3:e1487. https://doi.org/10.7717/peerj.1487.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41. https://doi.org/10.1093/nar/gks1219.
Guillou L, Bachar D, Audic S, Bass D, Berney C, Bittner L, et al. The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013;41:D597–D604.
Ratnasingham S, Hebert PDN. BOLD: The barcode of life data system: barcoding. Mol Ecol Notes. 2007;7:355–64.
Machida RJ, Leray M, Ho SL, Knowlton N. Metazoan mitochondrial gene sequence reference datasets for taxonomic assignment of environmental samples. Sci Data. 2017;4(January):1–7. https://doi.org/10.1038/sdata.2017.27.
Abarenkov K, Nilsson RH, Larsson KH, Alexander IJ, Eberhardt U, Erland S, et al. The UNITE database for molecular identification of fungi - recent updates and future perspectives. New Phytol. 2010;186:281–5.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Lanzén A, Jørgensen SL, Huson DH, Gorfer M, Grindhaug SH, Jonassen I, et al. CREST - classification resources for environmental sequence tags. PLoS One. 2012;7:e49334.
Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–7.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90. https://doi.org/10.1186/s40168-018-0470-z.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. https://doi.org/10.1093/bioinformatics/btq461.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Boyer F, Mercier C, Bonin A, Le Bras Y, Taberlet P, Coissac E. Obitools: a unix-inspired software package for DNA metabarcoding. Mol Ecol Resour. 2016;16:176–82.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. https://doi.org/10.7717/peerj.2584.
Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–10.
Esling P, Lejzerowicz F, Pawlowski J. Accurate multiplexing and filtering for high-throughput amplicon-sequencing. Nucleic Acids Res. 2015;43:2513–24. https://doi.org/10.1093/nar/gkv107.
Murali A, Bhargava A, Wright ES. IDTAXA : a novel approach for accurate taxonomic classification of microbiome sequences. Microbiome. 2018;6:1–14.
Frøslev TG, Kjøller R, Bruun HH, Ejrnæs R, Brunbjerg AK, Pietroni C, et al. Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nat Commun. 2017;8. https://doi.org/10.1038/s41467-017-01312-x.
Cordier T, Pawlowski J. BBI: an R package for the computation of benthic biotic indices from composition data. Metabarcoding Metagenomics. 2018;2:1–4.
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. https://doi.org/10.1186/1471-2105-13-31.
Kwon S, Lee B, Yoon S. CASPER: context-aware scheme for paired-end reads from high-throughput amplicon sequencing. BMC Bioinformatics. 2014;15.
Mahé F, Rognes T, Quince C, De Vargas C, Dunthorn M. Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ. 2015;3:e1420. https://doi.org/10.7717/peerj.1420.
Acknowledgements
We thank all the beta-testers for their patience during the first phase of the development and all of their useful feedbacks. We also thank Slim Chraïti for lending his name to the application.
Funding
This work was supported by the Swiss National Science Foundation grant 31003A \ _159709, and the Swiss Network of International Studies project “Monitoring marine biodiversity in the genomic era”.
Author information
Authors and Affiliations
Contributions
YD, FL, LAPG, JP and TC conceived the project. YD performed the core development and most of the module’s integration. TC contributed with module’s integration and some User Interface elements. YD, FL, LAPG and TC extensively tested the application. TC wrote the paper with input from all the authors. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dufresne, Y., Lejzerowicz, F., Perret-Gentil, L.A. et al. SLIM: a flexible web application for the reproducible processing of environmental DNA metabarcoding data. BMC Bioinformatics 20, 88 (2019). https://doi.org/10.1186/s12859-019-2663-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-019-2663-2