
Abstract
Background
High-throughput sequencing has made it theoretically possible to obtain high-quality de novo assembled genome sequences but in practice DNA extracts are often contaminated with sequences from other organisms. Currently, there are few existing methods for rigorously decontaminating eukaryotic assemblies. Those that do exist filter sequences based on nucleotide similarity to contaminants and risk eliminating sequences from the target organism.
Results
We introduce a novel application of an established machine learning method, a decision tree, that can rigorously classify sequences. The major strength of the decision tree is that it can take any measured feature as input and does not require a priori identification of significant descriptors. We use the decision tree to classify de novo assembled sequences and compare the method to published protocols.
Conclusions
A decision tree performs better than existing methods when classifying sequences in eukaryotic de novo assemblies. It is efficient, readily implemented, and accurately identifies target and contaminant sequences. Importantly, a decision tree can be used to classify sequences according to measured descriptors and has potentially many uses in distilling biological datasets.
Similar content being viewed by others

Background
Low-cost DNA sequencing, computing power and sophisticated assembly algorithms have made it possible to readily assemble genome sequences. However, most organisms do not live in sterile environments and extracted DNA may be contaminated with foreign DNA from associated microbiota [1–3] and endosymbionts [4]. Laboratory reagents and procedures can also introduce foreign DNA [5–7] and eliminating these sequences remains a challenge [8]. Contaminants end up sequenced and assembled along with the DNA of the target organism and, if not eliminated, will become part of the assembled genome sequence.
Contamination errors are frequent in public databases [9–11]. For example, Merchant et al. [10] identified microbial contamination in genome sequences of the cattle Bos taurus and an additional 50% of the publicly available genomes they analyzed. Contamination has also been reported in human [7, 12] and microbiome [6] sequences. Crisp et al. [11] analyzed horizontal gene transfer (HGT) in 40 metazoan genomes but excluded 9 from HGT analyses due to extensive contamination.
Contamination can mislead scientific studies. For example, contaminant sequences may be mistaken for HGT or complicate efforts to analyze HGT. In the Crisp study discussed above [11] genes initially classified as the result of HGT but later marked as probable contaminants had common characteristics. Sixty-nine of the nematode Caenorhabditis japonica HGT-derived genes were not physically linked to metazoan genes, lacked introns and were likely contaminants. A separate study [9] reported that several genes in the nematode C. angaria genome sequence were thought to be HGT-derived but analyses revealed 14% of the assembled genome was contributed by bacterial contaminants. Analyses of the sea anemone Nematostella vectensis genome [13] indicated a shikimic acid pathway not previously found in metazoans [14] but a later study found these genes were from proteobacteria ‘consorts’ and not the result of HGT [4]. The tardigrade Hypsibius dujardini genome was reported as 17% HGT-derived [15] but later analyses indicated large scale [16–18] contamination and an actual HGT-derived content of 1-2% [3].
Current decontamination methods eliminate known or well-characterized contaminants. For example, the software package DeconSeq filters sequences based on a contaminant database [19]. However, contaminants in de novo assembly projects are often not known. In this situation filtering methods must eliminate sequences based on nucleotide similarity to possible contaminants [20] or select target sequences based on similarity to known sequences in public databases [21]. Both of these approaches risk eliminating DNA from the target organism. For example, for eukaryotic genomes possible contaminants include large segments of bacteria, plants, fungi, viruses and archaea. The sheer number of possible sequences leaves filtering methods prone to ‘overfitting’ a model of contaminant identity as sequences from the target organism may resemble contaminants due to random chance. This is especially problematic when working with sequences from non-model organisms as there may be few representatives in public databases.
Conversely, sequences from the target organism may resemble contaminants because they result from true HGT. Aggressively eliminating these sequences can remove true HGT. For example, pre-assembly filtering for possible contaminants removed horizontally-transferred Wolbachia sequences from the first version of the Drosophila ananassae genome sequence [22]. Subsequent analysis and re-assembly revealed that > 1 Mb of the Wolbachia genome had been transferred into D. ananassae [22, 23].
Here, we introduce a novel application of a supervised machine learning method, a decision tree, for identifying target and contaminant DNA in de novo genome assembly projects. Supervised machine learning works by constructing a model from a set of training data and using this model to predict classification responses. Decision trees do not require data transformations or normalizations and produce simple, easily interpretable relationships. Their simplicity means they are well-suited for classifying data with straightforward but nonlinear relationships to predictors. Decision trees are well-established in machine learning but not commonly used in biology or bioinformatics.
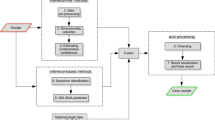
The majority of sequence filtering approaches have been developed for metagenomic datasets and an important question is whether methods developed for ‘binning’ microbial species can be co-opted for decontaminating eukaryotic genome sequences. For example, the frequency of short DNA ‘words’ of length k or k-mers can be used to classify microbes in metagenomic datasets [24–27]. Unsupervised classification methods bin samples based on sequence feature analysis (for example, [28, 29]) or combine sequence analysis with information on DNA sequencing coverage [30], taxonomy [31], and sequence composition [32]. Additionally, there are methods that employ both unsupervised and supervised methods to bin samples (for example, [33, 34]). Here, we evaluate the performance of our decision tree methods compared to the metagenomic classification software packages Anvi’o [32] (with CONCOCT [30] binning), Busybee [34] and Kraken [20] and the sequence filtering method Blobology [21] (Fig. 1).

The workflow from raw DNA sequence reads to assembled genome sequence for Anvi’o with CONCOCT binning, Busybee, Blobology, Kraken, and the decision tree. Both Blobology and Kraken required pre-assembly, filtering for target and contaminant reads, and final assembly. The decision tree, Anvi’o and Busybee filtered for target and contaminant scaffolds by constructing models and classifying contiguous sequences after assembly
We found that the decision tree accurately classified target and contaminant sequences based on measured descriptors. Importantly, the decision tree did not require a priori identification of significant descriptors and identified informative measures in constructing the model. Current decontamination methods can be time-consuming and require multiple manual steps that reduce reproducibility. In contrast, decision tree decontamination is readily implemented. The generality of the method means there are potentially many uses in biology.
Results
Genome sequences
We implemented our decision tree on three empirical datasets and twenty simulated datasets. The ‘real’ organisms included two nematodes from laboratory cultures, C. remanei and C. latens, that were found to be contaminated with microbes and one rotifer, Adineta vaga. The bdelloid rotifer A. vaga is both ameiotic and asexual and 8% of its genes are of non-metazoan origin [35]. We included A. vaga to determine if a decision tree could accurately separate foreign DNA from horizontally transferred DNA in an organism with high levels of confirmed HGT [35–37]. In order to test the methods on a range of genome-contaminant data structures we also simulated genomic and transcriptomic libraries from the published gene sequences of the plant Arabidopsis thaliana, the nematode C. elegans, the fruitfly D. melanogaster, and the pufferfish Takifugu rubripes. We contaminated each of these with a single microbe, the yeast Candida albicans, a low coverage mix of the microbial species listed above, an archaeon from the microbial dark matter project [38] and a mix of Homo sapiens and the common microbial contaminant Bradyrhizobium sp. [5, 6].
Prokaryotic contaminants in empirical genome sequences
The C. remanei genome sequence was estimated to be 131 Mb (Table 1) by flow-cytometry [39, 40] and initial analyses with Basic Local Alignment Search Tool (BLAST) [41] indicated that the assembly contained excess sequence due to microbial contaminants. The most prevalent taxonomic origin in the entire assembled genome set was C. remanei (Fig. 2) and the second most prevalent origin was the microbial contaminant E. coli. The third most prevalent organism was an unnamed Chryseobacterium species, also a microbial contaminant. 409 scaffolds could not be assigned taxonomic origin with BLAST.

The top 20 organisms identified in BLAST analysis of the empirical genome sequences for (a) C. remanei (b) C. latens (c) A. vaga. For C. remanei the most common BLAST hit was C. remanei, followed by two likely contaminants and scaffolds that could not be assigned origin with BLAST. For C. latens the most common BLAST hit was the microbial contaminant S. matophilia followed by C. remanei, a second contaminant P. protegens, and scaffolds that could not be assigned origin. For A. vaga the majority of scaffolds could not be assigned origin with BLAST, likely due to the low number of rotifer sequences in public databases
For C. latens the most prevalent taxonomic origin was a microbial contaminant, Stenotrophomonas maltophilia, that was also found in the C. remanei assembled sequence (Fig. 2). The second most prevalent taxonomic origin was C. remanei. This is likely because C. latens is a recently described species (previously C. species 23 [42]) and there are few C. latens sequences in public databases. C. remanei and C. latens are closely related and partially interfertile [43]. We were not able to identify a taxonomic origin for 429 of the assembled scaffolds.
For the A. vaga dataset there were non-metazoan BLAST alignments as expected under a model of high HGT. BLAST could not identify a taxonomic origin for 34,264 A. vaga scaffolds (Fig. 2) which was likely due to the low number of rotifer sequences in public databases. In order to identify probable contaminants we focused on an unusual pattern of 989 BLAST alignments to a single strain of E. coli (K-12 strain C3026), 206 BLAST alignments to a single strain of the human pathogen Shigella flexneri (4c), and 26 BLAST alignments to the microbe Variovorax paradoxus.
Identifying contaminants with predictor variables
We removed the target species genome sequences from the NCBI nucleotide (nt/nr) database and used BLAST to assign taxonomic origin. We aligned DNA and RNA sequence reads to each genome and calculated 8 predictor variables for scaffolds: (1) length, (2) GC content, (3) mean DNA sequencing coverage, (4) mean RNA sequencing coverage, (5) percent of scaffold covered in DNA alignment, (6) percent of scaffold covered in RNA alignment, (7) GC content of aligned DNA reads, and (8) GC content of aligned RNA reads.
We selected a portion of the scaffolds with BLAST-assigned taxonomy as a training set and used the remainder of scaffolds with BLAST-assigned taxonomy as a test dataset. We used the training set to construct a decision tree and used this tree to classify each of the test scaffolds as either target or contaminant. We varied the portion of the dataset used in training from 1-99% and calculated the mean and standard deviation of accuracy, sensitivity, and specificity across 100 replicates (results for C. remanei Fig. 3a). Here, model error was the percent of scaffolds in the test dataset that had a BLAST-assigned origin and were mis-classified. Accuracy was measured as 1-error. Sensitivity was calculated as T P/(T P+F N) where TP was the number of true positives and FN was the number of false negatives. Specificity was calculated as T N/(T N+F P) where TN was the number of true negatives and FP was the number of false positives. True positives were correctly identified target organism sequences and true negatives were correctly identified contaminants. Accuracy, sensitivity, and specificity plateaued with >40% of the data used for training (Figs. 3 and 4) and we used 50% of the dataset for decision tree training.

Accuracy, sensitivity and specificity for (a) decision tree and (b) bagging decision tree models. Decision tree models achieved high accuracy, sensitivity and specificity but were influenced by variation in the training dataset. The bagging decision tree model achieves high accuracy, sensitivity and specificity with lower variance between models constructed with different training datasets. For the decision tree models accuracy, sensitivity and specificity plateau with >25% of the data used in training while the performance of the bagging model plateaus with >40% of the data used in training

Accuracy, sensitivity and specificity for (a) random forest and (b) boosted decision tree models. Both random forest and boosted decision tree models resulted in high accuracy, sensitivity and specificity but showed non-monotonic responses to the training datasets
Decision trees are susceptible to bias and variance due to variation in the training dataset (Fig. 3a). In order to construct more accurate models we used a variation of a bootstrap procedure, bootstrap aggregation or ‘bagging’, that reduces the variance of the decision tree model (Fig. 3b). We also estimated the performance of random forest models (Fig. 4a) and boosted decision tree models (Fig. 4b). Accuracy, sensitivity and specificity were >99.5% for each of these models but the random forest and boosted models did not show monotonic responses to the proportion of data used in training and we used bagged decision tree models for the remainder of the analyses. Sensitivity exceeded specificity for all models (Figs. 4 and 5).

GC content and the average per-base sequencing coverage for individual scaffolds in the empirical datasets (a) C. remanei training; (b) C. remanei full dataset; (c) C. latens training; (d) C. latens full dataset; (e) A. vaga training; and (f) A. vaga full dataset. Training datasets with BLAST-identified origins are shown on the left and decision tree bagging model predictions for full datasets are shown on the right with model error
For C. remanei the bagging model predicted 19.38 Mb contained in 2470 scaffolds did not have a Caenorhabditis origin (Table 2; Fig. 5a-b). The contaminant sequences predominantly had low sequencing coverage (on average less than 10x; Fig. 5b) and GC content ranging from 35–70% or moderate sequencing coverage (on average, similar to that for scaffolds of Caenorhabditis origin) with high GC content (greater than 60%) although >50 scaffolds had GC/coverage profiles that deviated from this pattern. Of the 409 scaffolds without taxonomic origin the bagged decision tree model predicted 213 were contaminants.
For C. latens 17.06 Mb contained in 2896 scaffolds were of non-Caenorhaditis origin (Table 2; Fig. 5c-d). The model predicted that 28 of the 429 scaffolds without BLAST-identified origin were contaminants. The contaminant scaffolds had moderate-to-high sequencing coverage that actually exceeded the sequencing coverage of the C. latens scaffolds for roughly 1/3 of the contaminant scaffolds. The GC content of contaminant scaffolds was 55-70% while the GC content of the C. latens scaffolds was 30-50%.
The decision tree predicted 0.62 Mb contained in 2887 scaffolds were contaminants in the A. vaga genome sequence (Table 2; Fig. 5e-f). The model predicted 1593 of the 34,262 scaffolds without BLAST-identified taxonomy were contaminants. The contaminant scaffolds were small sequences with a median size of 59 bp and a mean size of 169 bp. In contrast, the true Adineta scaffolds had a median size of 408 bp and a mean size of 1080 bp. Contaminant scaffolds had GC content >40% while the Adineta scaffolds had GC content <45%.
Predictor variables
For each dataset we randomly selected 50% of the scaffolds with BLAST-assigned taxonomy as a training dataset and constructed bagged decision tree models for 2-8 variables. We repeated this procedure 1000 times and calculated the mean and standard deviation of accuracy, sensitivity, and specificity for each of these predictor combinations. Here, we focus on results for C. remanei (Fig. 6a). Mean DNA sequencing coverage and mean RNA sequencing coverage had the highest Gini importances and a model constructed solely with these predictors was able to correctly classify >97% of the C. remanei dataset. When a third predictor, the percent of the scaffold covered in RNA alignment, was added the model correctly classified >98% of the dataset. Model accuracy and sensitivity plateaued above 99.5% when a fourth variable, scaffold GC content, was included but specificity increased slightly as successive predictors were added to the model.

Accuracy, sensitivity and specificity for (a) the decision tree bagging model constructed with 2-8 predictors and (b) Anvi’o with CONCOCT binning and Busybee. Acccuracy and sensitivity for the decision tree bagging model plateau with 4 predictors but small increases in specificity resulted from additional predictors. Anvi’o had the highest specificity compared to the decision tree bagging model or Busybee while Busybee had the highest sensitivity
Software comparisons
We compared the decision tree bagging model results against those produced by Anvi’o [32] with CONCOCT binning [30], Busybee [34], Kraken [20] and Blobology [21]. Processing our sequencing files with Anvi’o was time-intensive and because of that we chose to proceed with the default setting and analyzed the 2304 scaffolds >2500 bp. We calculated accuracy, sensitivity and specificity based on this smaller scaffold set. Anvi’o [32] separated the contaminated C. remanei genome sequences into 18 bins however 3 of these contained only 1 scaffold. Seven bins contained primarily C. remanei sequences. Specificity was high (Fig. 6b) and Anvi’o misclassified just 2 Chryseobacterium scaffolds as Caenorhabditis. However, the overall Anvi’o accuracy rate was lower at 98.1% with 5 misclassified scaffolds and 38 scaffolds that were entirely unclassified. Of these, 21 were Caenorhabditis sequences and sensitivity was 98.5%.
Busybee [34] separated the contaminated C. remanei genome sequences into 5 bins. Busybee had a sensitivity rate of 99.89% (Fig. 6b) and placed just 2 Caenorhabditis scaffolds in microbial bins but the 2 Caenorhabditis bins (Fig. 7) contained 166 microbial scaffolds. Busybee bin 4 contained the majority of the C. remanei scaffolds with few microbial scaffolds (Fig. 7a) but Busybee bin 3 was a heterogeneous mix of scaffolds from C. remanei and Rhodococcus species (Fig. 7b).

Busybee bin 4 (a) contained primarily scaffolds of Caenorhabditis or unknown origin with few microbial contaminants while Busybee bin 3 (b) was a hetereogeneous mix of sequences with different origins. The scaffolds in Busybee bin 3 separated by taxonomic origin when visualized by scaffold GC content and sequencing coverage
Pre-assembly filtering methods can not be evaluated with accuracy, sensitivity and specificity and instead we measured the resulting genome size and genic completeness with BUSCO [44] and CEGMA [45]. BUSCO searches for a set of 982 orthologous genes thought to exist in single-copy in metazoans and CEGMA searches for a set of 248 ultra-conserved eukaryotic orthologous genes. For C. remanei the Blobology protocol [21] resulted in a genome sequence 0.75 Mb smaller than the decision tree genome sequence. We repeated the Blobology protocol focusing on a single contaminant order, Xanthomonadales, and assembled a complete genome sequence for the microbe S. maltophilia [46]. Using Kraken [20] for pre-assembly filtering resulted in a genome sequence 9.3 Mb shorter than the decision tree sequence. The decision tree assembled sequence contained a greater proportion of the BUSCO and CEGMA gene sets when compared with Blobology and Kraken (Table 3).
Identifying contaminant sequences in simulated genomes
We assembled the simulated libraries with low coverage microbial sequences, archaeons, and H. sapiens/Bradyrhizobium contaminants but BLAST failed to identify any scaffolds with these taxonomic origins in the resulting genome sequences. Accordingly, we focused on the simulated libraries with microbial and fungal contaminants for decision tree decontamination.
The simulated libraries with microbial contaminants were disentangled with decision tree models constructed solely on the scaffold GC content and the average per-base DNA sequencing coverage (Fig. 8). The simulated microbial contaminants had scaffold GC contents of 50-69% while the target organisms had scaffold GC contents of 24-72%. The GC content of the assembled C. albicans scaffolds ranged from 23-53% and was similar to the target organisms which had GC contents of 24-72% (Fig. 9). Accordingly, the C. albicans-contaminated simulated libraries showed poor discrimination with a decision tree model constructed with scaffold GC content and average per-base sequencing coverage (error rates >10%). For each simulated library contaminated with C. albicans we constructed a model with the full eight variables to increase prediction accuracy > 99% (Fig. 9).

GC content and average per-base sequencing coverage for the simulated datasets contaminated with microbial DNA. Training datasets are shown on the left and bagging decision tree predictions are shown on the right for a-b) A. thaliana; c-d) C. elegans; e-f) D. melanogaster; and g-h) T. rubripes. The microbial genomes were GC-rich relative to the target organisms and a simple decision tree based on GC content and sequencing coverage predicted scaffold origin with low error for each dataset

GC content and average per-base sequencing coverage for the simulated datasets contaminated with C. albicans DNA. Training datasets and bagging decision tree predictions are shown for a-b) A. thaliana; c-d) C. elegans; e-f) D. melanogaster; and g-h) T. rubripes. C. albicans and the target organisms had similar GC contents and the bagging decision tree predictions were based on a complex relationship that included multiple predictors and mRNA data
Discussion
We have developed a novel implementation of a decision tree, an established machine learning method, for distilling and decontaminating de novo assembled genome sequences. Our method filters based on any measurable characteristic. Here, we have focused on eight predictors and constructed decision tree models for empirical and simulated datasets. These models accurately predicted target or contaminant status for >99% of the scaffolds for which we could assign taxonomic origin with BLAST [41]. Importantly, we were able to classify scaffolds as target or contaminant in the absence of BLAST information based on predictor variables. Decision tree decontamination works on measurable sequence characteristics and is particularly useful for non-model organisms and those with low representation in public databases. Additionally, the influence of existing contamination in public databases can be limited by reducing training dataset size and manually curating training data.
Decontamination and dataset GC structure
In our model runs the complexity of the decision tree was influenced by the GC structure of the target and contaminant genome sequences. For example, the simulated datasets with bacterial contaminants were accurately decontaminated with a simple model based on scaffold GC content and average per-base sequencing coverage. Although genomic GC content varies broadly, metazoan genomes skew towards an enrichment of AT nucleotides while the GC content of bacterial genomes ranges from <15% to >75% [47]. In the simulated libraries these differences, coupled with discrete differences in the average per-base sequencing coverage, were large enough to accurately discriminate between target and contaminant sequences. These results indicate that discriminating between target and contaminant sequences in empirical datasets may be straightforward if the target and contaminant genomes have very different GC structures. For example, an easily discriminated case may be identifying sequences from a single high-GC contaminant in an invertebrate genome assembly.
The simulated libraries were created from high-quality genome sequences assembled with high certainty. Despite this, there was large variability in the estimated sequencing coverage (for example, Fig. 8). The ART [48] simulation software we used produces sequence reads according to a model based on real Illumina datasets and includes coverage variability and substitution, insertion and deletion errors. However, very large coverage values like the maximum sequencing coverage estimates we have reported here result in part from difficulties that arise in aligning relatively short 150 bp sequence reads to long repeats and other complex structures in metazoan genome sequences. Even in these ‘ideal’ simulated situations, the average per-base sequencing coverage did not reliably separate target and contaminant DNA sequences.
Including multiple predictor variables.
For our empirical datasets we were able to classify targets and contaminant sequences with relatively high accuracy (>90%) with decision tree models constructed solely on GC content and DNA sequencing coverage. However, achieving >99% accuracy, sensitivity and specificity required decision tree models constructed with at least 4 predictor variables. This was also true for the simulated datasets contaminated with the yeast C. albicans (Fig. 9).
The eight predictor variables we chose reflected different aspects of the assembly process and the biological basis of the sequences. For example, scaffold length is a measure of how reliably that segment of DNA assembled and depends on the sequencing coverage in the total sequence read library and the complexity of the sequence. In contrast, the percent of the scaffold covered in aligning RNA reads to the assembled scaffold measures gene density which varies by taxonomic origin. For example, the microbial P. aeruginosa genome is 89.4% coding sequence [49] while the genome of the vertebrate T. rubripes is 22.23% coding sequence [50]. Each of these descriptors had different Gini importances for the decision tree models constructed in this study and the minimal sets of predictor variables would likely differ for different target-contaminant combinations.
Software comparisons
We compared the C. remanei assembled sequences produced by our decision tree model with those produced by Anvi’o with CONCOCT binning [30, 32], Busybee [34], Blobology [21] and Kraken [20]. Both Anvi’o with CONCOCT binning and Busybee were developed for disentangling metagenomic datasets and microbial sequences. Anvi’o excelled at identifying microbial sequences but the decision tree model had higher accuracy and sensitivity than Anvi’o when constructed with >3 predictor variables. Busybee had an extremely high sensitivity but lower accuracy and specificity relative to both the decision tree models and Anvi’o. Metagenomic tools that are tuned for success with microbial sequences may not be capable of achieving the same levels of success in decontaminating eukaryotic datasets.
Anvi’o was time-intensive in pre-processing and analyzing sequences and could not accurately classify scaffolds < 2500 bp. In comparison, Busybee [34] was readily available through a web portal and accurately classified >95% of the complete C. remanei scaffold set in less than 10 minutes. The time-limiting step in decision tree decontamination is aligning the DNA and RNA reads to the assembled genome. Once this is complete model construction and scaffold selection can be completed within minutes.
Kraken overfit a model of contaminant identity and the assembled C. remanei sequence was shorter and had the smallest complement of orthologous genes [44] when compared with either the decision tree or Blobology sequences. Blobology resulted in an assembled sequence that was 99% of the length of the decision tree assembled sequence. The decision tree sequence had a greater percentage of orthologous genes [44, 45] when compared with both the Blobology and Kraken sequence. These results indicate that decontaminating and producing a complete assembled genome may require tools that are specifically tuned for certain scenarios. For example, the C. remanei sequence contained at least one full bacterial genome [46]. The raw ALLPATHS-LG [51] assembly contained many partial bacterial sequences but Blobology’s targeted filtering and re-assembly produced a high-quality sequence for the contaminant Stenotrophomonas maltophilia.
Classification errors
We began this study motivated by the goal of decontaminating assembled genome sequences without removing HGT. Under this conceptual framework we might expect that classification errors relate to horizontally transferred sequences but we did not find this. Each method produced a small number of errors and we were able to investigate these individually. For example, 2 scaffolds in the C. remanei dataset were predicted as contaminants by the decision tree model but were identified as related to the nematode Parastronglyoides by BLAST. Although Parastronglyoides is a nematode, the full list of BLAST hits for these sequences included multiple microbes and no other nematodes. One scaffold was binned with E. coli sequences by both Anvi’o [32] and Busybee [34] and although BLAST identified it as related to the nematode C. elegans, the full list of BLAST hits for this sequence included multiple microbes as well. The BLAST-assigned taxonomic identity for some of these errors may reflect contamination in public databases.
Other mis-classifications did not reflect contamination errors. Anvi’o [32] failed to classify two scaffolds that did not express mRNA but that BLAST identified as closely related to C. elegans. These scaffolds contained gene sequences encoding srh-266, a serpentine or chemoreceptor, and a glycosyltransferase with conserved single copy homologs across Caenorhabditis, Drosophila and Danio rerio. Busybee [34] binned several megabases of Rhodococcus sequence and a Rhodococcus plasmid with Caenorhabditis sequences including a 137,654 bp scaffold that expressed mRNA and contained a gene sequence encoding a homolog to C. elegans fibrillin-1, an extracellular matrix protein with a human homolog that results in Marfans syndrome when mutated.
These sequences did not have any readily observable patterns and were likely binned together because of similarities in k-mer frequencies. We do not understand how mutation and functional convergence influence the evolution of genome sequences. Complete knowledge of supervised and unsupervised sequence analysis methods will require a deeper understanding of the rules that govern change at a genomic level.
Conclusions
Here, we have presented a novel implementation of a decision tree model for decontaminating de novo sequence assemblies. Our method is readily implemented, reproducible and fast. We have shown that it can rigorously decontaminate sequences and is useful for non-model organisms. Machine learning methods are established in other disciplines but not yet common in biology. We hope that this example demonstrates the utility of machine learning methods for distilling meaning in biological datasets.
Methods
Empirical genome sequences
Genomic DNA was isolated from C. remanei and C. latens nematode worms (for detailed experimental methods, see [52]). We sequenced one overlapping paired-end genomic library with an average fragment size of 180bp (as required by the assembly software ALLPATHS-LG [51]) for each nematode. We sequenced three mate pair genomic libraries with average fragment sizes of 0.7-2, 2-4, and 4-7 kb for C. remanei and three mate pair libraries with average fragment sizes of 4-6 kb, 6-9 kb and 9-12 kb for C. latens. Libraries were sequenced as 2 x 101nt reads with an Illumina HiSeq. We sequenced these libraries at high depth and in order to avoid biased errors we used the k-mer filter protocol in the software package Stacks [53] to pre-filter the overlapping paired end fragments by k-mer frequency spectra. We used k=15 and removed reads with greater than 12 rare k-mers (single occurrences) and greater than 51 abundant k-mers (here, defined as 20,000 or more occurrences). We used the ALLPATHS-LG (version 52488) [51] genome assembly software package which performs k-mer spectra correction of sequencing errors (with k=25), builds contiguous sequences with a de Bruijn graph from the 180bp reads, and constructs scaffolds with mate pair libraries.
We isolated total RNA from mixed-stage populations of C. remanei and C. latens, purified mRNA, synthesized cDNA libraries and sequenced these libraries as 2 x 101nt reads with an Illumina HiSeq. We used the MAKER2 software package [54] to annotate protein-coding genes (for detailed methods see [52]).
The A. vaga sequence read libraries were obtained from the Sequence Read Archive [55] and assembled genome sequences were obtained from the National Center for Biotechnology Information (NCBI) [56]. For details on DNA and RNA isolation, sequencing, assembly and annotation see [35].
Sequencing the C. remanei paired-end genomic library produced 367,673,013 overlapping pairs (statistics for C. remanei libraries are given in Additional file 1: Table S2). We removed 75,850,115 sequence reads with rare k-mers and 22,433,210 sequence reads with abundant k-mers resulting in 637,062,701 retained reads. Sequencing the C. latens paired-end library produced 171,027,578 overlapping pairs (C. latens library statistics are given in Additional file 1: Table S3). We removed 46,491,040 reads with rare k-mers and 11,359,209 reads with abundant k-mers resulting in 284,204,907 retained sequence reads. We filtered the sequenced mRNA libraries for adapter contamination and retained 26,170,962 C. remanei reads and 32,459,744 C. latens reads for transcript assembly. Statistics for the A. vaga libraries used in this study are given in Additional file 1: Table S4 and statistics for the simulated genomic and transcriptomic libraries are given in Additional file 1: Tables S5 and S6.
Simulated genome sequences
We simulated DNA sequence reads for A. thaliana, C. elegans, D. melanogaster and T. rubripes and contaminated these libraries with sequences from Agrobacterium radiobacter, Pseudomonas aeruginosa, Escherichia coli, Ralstonia sp. 5_7_47FAA [6], C. albicans, microbial dark matter archaea [38], Homo sapiens and the common microbial contaminant B. sp. BTAi1 [5, 6]. Due to the large size of the human genome [57] we simulated sequences from the H. sapiens mitochondrial chromosome and chromosome X, Y, IV, XII, and XX. GenBank accessions for genome sequences are listed in Additional file 1: Table S1.
We simulated Illumina sequence read datasets with the software package ART (version ART-MountRainier-2016-06-05) [48]. We generated two genomic DNA libraries for each target organism, one a 150 bp paired-end library with a 270 bp fragment, standard deviation of 30 bp (resulting in, on average, 10% overlap between the paired reads) and average per-base sequencing coverage of 100x and one a 150 bp mate pair library with a 2500 bp fragment, standard deviation of 50 bp and average per-base sequencing coverage of 33x. We also generated two genomic DNA libraries for each contaminant organism with the parameters listed above but lower average per-base sequencing coverage. For the work reported here, the contaminant paired-end sequencing coverage was 20x and the mate pair sequencing coverage was 10x. We combined target and contaminant to create contaminated libraries and assembled genome sequences with ALLPATHS-LG [51].
We also simulated RNA libraries with the software package ART [48]. For each target organism we simulated a 100bp paired-end library with a 400bp fragment, standard deviation of 50 bp and an average sequence coverage of 30x. For each contaminant organism we simulated a library with the parameters listed above but a lower sequencing coverage of 10x.
Assigning taxonomic identity to scaffolds
We used the Basic Local Alignment Search Tool (BLAST 2.3.1+) to identify the single best BLASTn [41] match (expect threshold=10; word size=28; match/ mismatch scores=1,-2, gap costs=linear) for each assembled scaffold. We used the NCBI nt database and for each target organism we removed that species genome sequences from the database. For the C. remanei and C. latens datasets we filtered these by genus and assigned each scaffold a taxonomic identity of ‘Caenorhabditis’, not ‘Caenorhabditis’ or ‘No BLAST hit.’ Note that some of the scaffolds identified in this way may actually be matching residual contamination in other Caenorhabditis assemblies (for example, the suspected contaminants in C. angaria [9] and C. japonica [11]). This may contribute to error in our datasets but we chose to maintain consistency with published results.
BLAST could not assign taxonomic identity for 34,264 A. vaga scaffolds (88% of the total scaffolds; 35.5 Mb of assembled sequence; 16.3% of the genome) as expected for a non-model organism. We assigned the 34,264 unidentifiable scaffolds a taxonomic identity ‘No BLAST hit,’ the E. coli K-12 strain C3026, S. flexneri 4c and V. paradoxus scaffolds a taxonomic identity of ‘Not Adineta’ and the remaining scaffolds a taxonomic identity of ‘Adineta.’
We classified the simulated assembled scaffolds by genus with BLAST with the exception of the T. rubripes datasets. Each of these contained scaffolds that BLAST could not assign taxonomic identity to and scaffolds that aligned to other metazoans. In order to focus on contaminants we assigned each scaffold a taxonomic identity of ‘No BLAST hit,’ ‘Other Animalia,’ ‘Takifugu,’ or the identified contaminant.
Calculating descriptors for training scaffolds
We used the program GMAP-GSNAP (version 2017-03-17) [58] to align DNA and RNA sequence reads to each assembled genome sequence. Reads were mapped as paired, correct orientation was required and we selected the single highest scoring location (no multimapping was allowed). Mate pair libraries may contain chimeric sequences and other artifacts that would align incorrectly and we eliminated these from coverage calculations. We used the BBMap/BBTools software package (version 37.32) [59] to calculate the length and GC content of each assembled scaffold. We also calculated the average sequencing fold coverage across each scaffold, the percent of scaffold covered in aligning reads to the assembled sequence and the GC content of the reads aligned to the assembled sequence for both DNA and RNA libraries.
The decision tree algorithm
The decision tree framework measured the information gain contributed by each variable according to the entropy H(S)=−Σ x∈X p x l o g 2 p x where H(S) was the entropy of dataset S, X were the classes in S and p(x) was the probability of x or the proportion of x in the dataset. Information gain was computed as I G(A,S)=H(S)−Σ t∈T p(t)H(t) where I G(A,S) was the difference in entropy from splitting on the variable or attribute A, T were subsets of S and p(t) was the proportion of t. Importantly, the decision tree framework did not require assumptions regarding which variables were significant and could classify data based on any measurable feature.
Data were classed according to Gini’s diversity index where \(G = \Sigma _{i}^{n_{c}} p_{i} (1- p_{i})\). Here, n c was the number of classes and and p(i) was the observed fraction of class i observations in the set. Training data was used to identify useful descriptors and construct a binary decision tree according to \( \widehat{y}=\underset{y=1,\dots, K}{\arg\ \min }{\varSigma}_{k=1}^K\widehat{P}\left(k|x\right)C\left(y|k\right). \) Here, \(\hat {y}\) was predicted classification, K the number of classes, \(\hat {P}(k|x)\) posterior probabilities, C(y|k) the cost of misclassification and arg min the input that minimized the value of the function.
We constructed bootstrapped aggregated or ‘bagged’ decision tree models by generating B different bootstrapped training datasets and averaging the predictions to obtain \(\hat {f}_{bag}(x) = \frac {1} {B} \sum ^{B}_{b=1}\hat {f}^{*b}(x)\phantom {\dot {i}\!}\) where \(\hat {f}^{*b}(x)\) is the set of predictions obtained from the bth bootstrap training set. We measured the Gini importance of each variable in the model as \(I = G_{parent} - G_{node_{1}} - G_{node_{2}}\phantom {\dot {i}\!}\), where G is the diversity index defined above. We also implemented a random forest model by bootstrapping training datasets and constructing decision trees from a random sample m of the p predictor variables. Here, m=4 and we constructed 5000 trees per model run. A random forest model is an extension of a bagging model and the two are equivalent when m=p. We implemented a boosted model by fitting decision trees to the residuals of a shallow tree and summing across these trees. Here, we allowed 2 splits in each tree, used a shrinkage parameter of 0.01 and fit 10 trees per model run.
Implementing Anvi’o with CONCOCT binning
We used the assembled genome sequence to create an Anvi’o (v2.4.0 “Pyrenees”) contig database with k-mer frequency (computed at k=4), GC content, open reading frames and predicted bacterial single-copy core genes for each scaffold [32]. We processed scaffolds > 2500 bp with anvi’o to estimate sequencing coverage profiles (mean, standard deviation and average coverage for inner quartiles) and characterize single-nucleotide variants for our DNA and RNA sequences. We used CONCOCT (v0.4.0) [30] for binning within Anvi’o. Briefly, CONCOCT bins metagenomic samples by analyzing both k-mer frequency and sequencing coverage across assembled scaffolds.
Implementing Busybee
We uploaded the assembled genome sequence as a.fasta to the Busybee (version 2017-01-09) [34] web portal (https://ccb-microbe.cs.uni-saarland.de/busybee). We enabled both taxonomic and functional annotation and uploaded our set of 8 predictor variables as a custom annotation file. We set the minimum contig length at 1000 bp, the minimum contig length for border points at 1000, the minimum contig length for cluster points at 2000, the k-mer length at 5, the probability at 0.0, the minimum points in neighbourhood at 30, and the transformation at standard.
Implementing Blobology
We implemented the Blobology protocol [21] to compare with decision tree decontamination. Briefly, a preliminary assembly was performed using ABySS-PE (version 1.9.0) [60]. For the C. latens and C. remanei datasets, only the standard paired-end data was assembled. The source FASTQs were aligned back to this new assembly using GMAP-GSNAP [58], and the assembly was classified using NCBI BLAST [41]. The BLAST results, assembly, and alignment were analyzed with blobtools (v0.9.19) [21]. The resulting TAGC plot was used to determine filtering conditions based on the GC content and average per-base sequencing coverage of each assembled contiguous sequence (GC<0.45 and coverage>2 for A.vaga, GC<0.45 for C. latens, and three passes for C. remanei [GC<0.65, GC<0.6 && coverage>11, GC<0.5 && coverage >5]). A list of contiguous sequences which met these conditions was generated, and paired-end and mate pair reads matching these contiguous sequences were selected using custom scripts alongside standard GNU tools and samtools [61]. The post-filtering reads were assembled with ABySS-PE [60] or ALLPATHS-LG [51]. Assemblies were evaluated using QUAST (version 4.5) [62].
Implementing Kraken
We also filtered our empirical assemblies with Kraken (version 0.10.5-beta)[20] to compare with the Decision Tree. Briefly, a preliminary assembly was performed using ABySS [60], and the source FASTQs were aligned to this assembly using GMAP-GSNAP [58]. Each sequence in this assembly was classified using Kraken [20]. Contiguous sequences successfully classified by Kraken were assumed to be contaminants due to the content of Kraken’s standard database (generated from NCBI FTP on April 4, 2016). This list was used to generate a list of contiguous sequences to keep from the data, and reads matching those contiguous sequences were selected using custom scripts alongside standard GNU tools and samtools [61]. The post-filtering reads were assembled with ABySS [60] or ALLPATHS-LG [51]. We used QUAST (version 4.5) [62], BUSCO (v2.0) with the nematodea_odb9 database [44] and CEGMA (version 2.4) [45] to assess the completeness of the decision tree, Blobology, and Kraken assemblies.
References
Kumar S, Blaxter ML. Simultaneous genome sequencing of symbionts and their hosts. Symbiosis. 2012; 55(3):119–26.
Artamanova II, Lappi T, Zudina L, Mushegian AR. Prokaryotic genes in eukaryotic genome sequences: when to infer horizontal gene transfer and when to suspect an actual microbe. Environ Microbiol. 2015; 17(7):2203–8.
Koutsovoulos G, Kumar S, Laetsch DR, Stevens L, Daub J, Conlon C, et al.No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proc Natl Acad Sci. 2016; 113:5053–8.
Artamanova II, Mushegian AR. Genome seuqence analysis indicates that the model eukaryotic Nematostella vectensis harbors bacterial consorts. Appl Environ Microbiol. 2013; 79(22):6868–73.
Laurence M, Hatzis C, Brash DE. Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS ONE. 2014; 9(5):e97876.
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al.Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology. 2014; 12:87.
Strong MJ, Xu G, Morici L, Splinter Bon-Durant S, Badoo M, Lin Z, et al. Microbial contamination in next generation sequencing: Implications for sequence-based analysis of clinical samples. PLoS Pathogens. 2014; 10(11):e1004437.
Ekblom R, Wolf JBW. A field guide to whole-genome sequencing, assembly and annotation. Evol Appl. 2014; 7:1026–42.
Percudani R. A microbial metagnome (Leucobacter sp.) in Caenorhabditis whole genome sequences. Bioinforma Biol Insights. 2013; 7:55–72.
Merchant S, Wood DE, Salzberg SL. Unexpected cross-species contamination in genome sequencing projects. PeerJ. 2014; 2:e675.
Crisp A, Boschetti C, Perry M, Tunnacliffe A, Micklem G. Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes. Genome Biol. 2015; 16:50.
Riley DR, SIeber KB, Robinson KM, White JR, Ganesan A, Nourbakhsh S, et al. Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples. PLoS Comput Biol. 2013; 9(6):e1003107.
Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, et al.Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007; 317:86–94.
Starcevic A, Akthar S, Dunlap WC, Shick JM, Hranueli D, Cullum J, et al.Enzymes of the shikimic acid pathway encoded in the genome of a basal metazoan, Nematostella vectensis, have microbial origins. Proc Natl Acad Sci U S A. 2008; 105:2533–7.
Boothby TC, Tenlen JR, Smith FW, Wang JR, Patanella KA, Osborne Nishimura E, et al.Evidence for horizontal gene transfer from the draft genome of a tardigrade. Proc Natl Acad Sci U S A. 2015; 112(52):15976–81.
Arakawa K. No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proc Natl Acad Sci U S A. 2016; 113(22):E3057.
Bemm F, Weiss CL, Schultz J, Forster F. Genome of a tardigrade: Horizontal gene transfer or bacterial contaminationProc Natl Acad Sci U S A. 2016; 113(22):E3054—6.
Delmont TO, Eren AM. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome asemblies. PeerJ. 2016; 4:e1839.
Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE. 2011; 6(3):e17288.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014; 15:R46.
Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. Blobology: exploring raw genome data for contaminants, symbionts, and parasites using taxon-annotated GC-coverage plots. Front Genet. 2013; 4(237):1–12.
Dunning Hotopp JC, Clark ME, Oliveria DCSG, Foster JM, Fischer P, Munoz Torres MC, et al.Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007; 317:1753–5.
Dunning Hotopp JC. Horizontal gene transfer between bacteria and animals. Trends Genet. 2011; 27(4):157–63.
Sandberg R, Winberg G, Branden CI, Kaske A, Ernberg I, Coster J. Capturing whole-gehome characteristics in short sequences using a naive Bayesian classifier. Genome Res. 2001; 11(8):1404–9.
DIck GJ, ANdersson AF, Baker BJ, Simmons SL, THomas BC, Yelton AP, et al.Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009; 10:R85.
Kislyuk A, Bhatnagar S, Dushoff J, Weitz JS. Unsupervised statistical clustering of environmental shotgun sequences. BMC Bioinformatics. 2009; 10:316.
Strous M, Kraft B, Bisdorf R, Tegetmeyer HE. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front Microbiol. 2012; 3:410.
Liao R, Zhang R, Guan J, Zhou S. A new unsupervised binning approach for metagenomic sequences based on N-grams and automatic feature weighting. IEEE/ACM Trans Comput Biol Bioinforma. 2014; 11(1):42–54.
Zhang R, Cheng Z, Guan J, Zhou S. Exploiting topic modeling to boost metagenomic reads binning. BMC Bioinformatics. 2015; 16(Supplement 5):S2.
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al.Binning metagenomic contigs by coverage and composition. Nat Methods. 2014; 11:1144–6.
Wang Y, Leung HCM, Yiu SM, Chin FYL. MetaCluster-TA: taxonomic annotation for metagenomic data based on assembly-assisted binning. BMC Genomics. 2014; 15(Supplement 1):S12.
Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, et al.Anvi‘o: an advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:e1319.
Kelley DR, Salzberg SL. Clustering metagenomic sequences with interpolated Markov models. BMC Bioinformatics. 2010; 11:544.
Laczny CC, Kiefer C, Galata V, Fehlmann T, Backes C, Keller A. BusyBee Web: metagenomic data anlysis by bootstrapped supervised binning and annotation. Nucleic Acids Res. 2017; 45(W1):W171–9.
Flot JF, Hespeels B, Li X, NOel B, Arkhipova I, Danchin EG, et al.Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature. 2013; 500(7463):453–7.
Gladyshev EA, Meselson M, Arkhipova IR. Massive Horizontal Gene Transfer in Bdelloid Rotifers. Science. 2008; 320(5880):1210–13.
Boschetti C, Carr A, Crisp A, Eyres I, Wang-Koh Y, Lubzens E, et al. Biochemical diversification through foreign gene expression in bdelloid rotifers. PLoS Genet. 2012; 8(11):e1003035.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J, et al.Insights into the Phylogeny and Coding Potential of Microbial Dark Matter. Nature. 2013; 499:431–7.
Bird DM, Blaxter ML, McCarter JP, Mitreva M, Sternberg PW, Thomas WK. A White Paper on Nematode Comparative Genomics. J Nematol. 2005; 37(4):408–16.
Haag ES, Chamberlin H, Coghlan A, Fitch DH, Peters AD, Schulenburg H. Caenorhabditis evolution: if they all look alike, you aren’t looking hard enough. Trends Genet. 2007; 23(3):101–4.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990; 215:403–10.
Felix MA, Braendle C, Cutter AD. A Streamlined System for Species Diagnosis in Caenorhabditis (Nematoda: Rhabditidae) with Name Designations for 15 Distinct Biological Species. Plos One. 2014; 9(4):e94723.
Dey A, Jin Q, Chen Y, Cutter AD. Gonad morphogenesis defects drive hybrid male sterility in asymmetric hybrid breakdown of Caenorhabditis nematodes. Evol Dev. 2014; 16(6):362–72.
Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015; 31(19):3210–2.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007; 23(9):1061–7.
Fierst JL, Murdock DA, Thanthiriwatte C, Willis JH, Phillips PC. Metagenome-assembled draft genome sequence of a novel microbial Stenotrophomonas maltophilia strain isolated from Caenorhabditis remanei tissue. Genome Announc. 2017; 5(7):e01646–16.
Li X, Du D. Variation, evolution, and correlation analysis of C+G content and genome or chromosome size in different kingdoms and phyla. PLoS ONE. 2014; 9(2):e88339.
Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics. 2012; 28(4):593–4.
Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al.Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000; 406:959–64.
Aparicio S, Chapman J, Stupka E, Putnam N, Chia Jm Dehal P, et al.Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu rubripes. Science. 2002; 297(5585):1301–10.
Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al.High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci. 2011; 108(4):1513–8.
Fierst JL, Willis JH, Tomas CG, Wang W, Reynolds RM, Ahearnge TE, et al. Reproductive mode and th evolution of genome size and structure in Caenorhabditis nematodes. PLoS Genet. 2015; 11(6):e1005323.
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: Building and Genotyping Loci de novo From Short-read Sequences. G3. 2011; 1:171–82.
Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011; 12:491.
Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011; 39:D19—21.
Pruitt K, Brown G, Tatusova T, et al. The Reference Sequence (RefSeq) Database 2002 Oct 9 [Updated 2012 Apr 6] In: McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda: National Center for Biotechnology Information (US); 2002. Chapter 18. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21091/.
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921.
Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005; 21(9):1859–75.
Bushnell B. BBMap: A fast, accurate, splice-aware aligner. 2800. Mitchell Drive, Walnut, Creek, USA: LBNL Department of Energy Joint Genome Institute; 2014. LBNL-7065E.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009; 19:1117–23.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al.The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009; 25:2078–9.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013; 29(8):1072–5.
Welch Mark DB, Meselson M. Oocyte nuclear DNA content and GC proportion in rotifers of the anciently asexual Class Bdelloidea. Biol J Linn Soc. 2003; 79:85–91.
Meyerowitz EM, Somerville CR. Arabidopsis. Cold Spring Harbor. New York: Cold Spring Harbor Laboratory Press; 1994.
The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000; 408:796–815.
Sulston JE, Brenner S. The DNA of Caenorhabditis elegans. Genetics. 1974; 77:95–104.
C elegans Sequencing Consortium. Genome sequence of the nematode C. elegans. Science. 1998; 282(5396):2012–8.
Laird CD, McCarthy BJ. Molecular characterization of the Drosophila genome. Genetics. 1969; 63:865–82.
Myers EW, SUtton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, et al.A whole-genome assembly of Drosophila. Science. 2000; 287:2196–204.
Brenner S, Elgar G, Sanford R, Macrae A, Venkatesh B, Aparicio S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature. 1993; 366:265–8.
Slater SC, Goldman BS, Goodner B, Setubal JC, Farrand SK, Nester EW, et al.Genome sequences of three Agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J Bacteriol. 2009; 191(8):2501–11.
Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, et al.The diploid genome sequence of Candida albicans. Proc Natl Acad Sci. 2004; 101(19):7329–34.
Blattner FR, Plunkett IIIG, Bloch CA, Perna NT, BUrland V, Riley M, et al.The complete genome sequence of Escherichia coli K–12. Science. 1997; 277(5331):1453–62.
Acknowledgements
JH Willis generated the Caenorhabditis empirical libraries. PC Phillips and AD Cutter provided helpful feedback and encouragement in developing the decision tree modeling. Mark Blaxter and an anonymous reviewer provided useful comments and direction in reviewing the manuscript.
Funding
This work was funded by new faculty startup funds from the College of Arts and Sciences at the University of Alabama and NIH R01 GM102511.
Availability of data and materials
Supplementary figures and tables are available with the online version of this article. The scripts used in this work, datasets and an example decision tree model are available at https://github.com/BamaComputationalBiology/DecisionTree. SIDR (Sequence Identification with Decision tRees) is a developing software package implementing these methods and available at https://github.com/damurdock/SIDR.
Author information
Authors and Affiliations
Contributions
JLF designed the study, performed the decision tree modeling, and wrote the article. DAM performed the Blobology, Kraken, CEGMA and BUSCO analyses. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1
Supplement: Fierst and Murdock, Decontaminating eukaryotic genome assemblies with machine learning. (PDF 9713 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fierst, J.L., Murdock, D.A. Decontaminating eukaryotic genome assemblies with machine learning. BMC Bioinformatics 18, 533 (2017). https://doi.org/10.1186/s12859-017-1941-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-017-1941-0