
Abstract
CD1d is a non-classical major histocompatibility class 1-like molecule which primarily presents either microbial or endogenous glycolipid antigens to T cells involved in innate immunity. Natural killer T (NKT) cells and a subpopulation of γδ T cells expressing the Vγ4 T cell receptor (TCR) recognize CD1d. NKT and Vγ4 T cells function in the innate immune response via rapid activation subsequent to infection and secrete large quantities of cytokines that both help control infection and modulate the developing adaptive immune response. T regulatory cells represent one cell population impacted by both NKT and Vγ4 T cells. This review discusses the evidence that NKT cells promote T regulatory cell activation both through direct interaction of NKT cell and dendritic cells and through NKT cell secretion of large amounts of TGFβ, IL-10 and IL-2. Recent studies have shown that CD1d-restricted Vγ4 T cells, in contrast to NKT cells, selectively kill T regulatory cells through a caspase-dependent mechanism. Vγ4 T cell elimination of the T regulatory cell population allows activation of autoimmune CD8+ effector cells leading to severe cardiac injury in a coxsackievirus B3 (CVB3) myocarditis model in mice. CD1d-restricted immunity can therefore lead to either immunosuppression or autoimmunity depending upon the type of innate effector dominating during the infection.
Similar content being viewed by others

Introduction
Myocarditis is an inflammation of myocardium with subsequent cardiomyocyte death, replacement fibrosis, and cardiac dysfunction [1, 2], is a significant cause of sudden death in children and young adults [3–7], and often follows cardiac infections (virus, bacteria, fungus, worms) [8]. Enteroviruses and adenoviruses cause approximately 80% of clinical viral myocarditis with human cytomegalovirus, parvovirus, influenza virus, and herpes simplex virus infection causing most of the remainder [9]. Cardiac injury results from direct viral injury to infected cardiocytes and from the host response to infection[10]. Strong evidence exists for immunopathogenic mechanisms of cardiac injury in experimental models of coxsackievirus B3 (CVB3) induced myocarditis. T cell depletion of mice dramatically reduces animal mortality and cardiac inflammation [11], and heart-specific, autoimmune CD8+ T cells isolated from CVB3 infected mice [12] transfer myocarditis into uninfected recipients. A major question is how the virus activates these autoimmune CD8+ T cells. Antigenic mimicry between CVB3 and cardiac myosin forms the basis for the autoimmunity [13, 14]. However, some CVB3 variants replicate in the heart but fail to activate autoimmunity [15]. The crucial difference between the variants is that the pathogenic virus induces CD1d up-regulation on hemopoietic and non-hemopoietic cells but the non-pathogenic variant does not [16–18], and this failure to up-regulate CD1d leads to generation of T regulatory cells [19].
CD1 molecules and regulation of their expression
CD1 molecules belong to a family of non-polymorphic, class I-like major histocompatibility complex (MHC) molecules, which bind and present amphiphilic lipid antigens to T cells for recognition [20]. The CD1 family in humans and most other species are divided into transmembrane Group 1 (CD1a,b,c), and Group 2 (CD1d) molecules [21, 22]. An intermediate isoform (CD1e) exists as a soluble molecule in the late endosome where it facilitates processing of complex glycolipids for presentation by other CD1 isoforms [23]. Group 1 CD1 molecules are expressed on thymocytes, dendritic cells, activated monocytes and B lymphocytes. CD1d is expressed on these cells and additionally on T cells and non-hemopoietic cells including cardiac myocytes and endothelial cells [16, 22, 24]. While structurally similar to class I MHC molecules (consisting of a single polypeptide chain coded by the CD1 gene and associated with β2 microglobulin), antigen presentation resembles class II MHC molecules since antigen loading occurs in the endosome pathway and is TAP independent [25–27]. The CD1 extracellular domain contains a deep antigen binding groove comprised of up to four hydrophobic pockets into which lipid tails of antigens are inserted [28–30]. CD1b presents bacterial lipids including mycobacterial mycolic acids [31], lipoarabinomannan [32], glucose monomycolate [33], and self-glycosphingolipids such as GM1 ganglioside [34]. CD1a and CD1c present bacterial phospholipids [35]. CD1d presents a bacterial sphingolipid from Sphingomonas[36], alphaproteobacterium from N. aromaticivorans[37], glycolipids from B. burgdorferi[38], and a self-sphingolipid isogloboside [39]. The sphingolipid α-galactosylceramide (αGalCer) isolated from marine sponges, is the classical CD1d ligand for activating NKT cells [40]. CD1 molecules also bind and present other endogenous (self) glycolipid sulfatides [41–44]. Lysosomal α-galactosidase A is highly effective in degrading endogenous lipid antigens, normally limiting autoreactive NKT cell responses [44]. However, infections inhibit α-galactosidase A activity allowing endogenous lipid accumulation and NKT cell activation. This means that CD1d dependent innate immunity may be directed to both exogenous and endogenous antigens during infections.
Group 1 CD1 molecules are not constitutively expressed on myeloid precursors of dendritic cells, but can be induced by signaling through TLR2 [45], TLR2/TLR5 agonists, or cytokines (GM-CSF and IL-4) during differentiation into immature dendritic cells [46]. CD1d is constitutively expressed in dendritic cells at most stages of differentiation, as well as on monocytes and macrophage [47]. Unlike Group 1 CD1 molecules, CD1d is not up-regulated by GM-CSF and IL-4 [24, 48], but is up-regulated by exposure to bacteria or viruses [16, 49, 50]. Studies using M. tuberculosis found that both signaling through TLR2 and cytokines (IFNγ and TNFα) were required for CD1d up-regulation on macrophage both in vitro and in vivo [51]. Similarly, studies using L. monocytogenes found that IFNβ increases CD1d expression [52]. CVB3 infection augments CD1d expression on macrophage, dendritic cells and T cells [18]. The virus also causes de novo CD1d expression on non-hemopoietic cells (cardiac endothelial cells and myocytes), but only in non-hemopoietic cells actively replicating virus. Immediately adjacent uninfected myocytes/endothelial cells remain CD1d negative [16]. CD1d expression requires TNFα, but TNFα treatment of uninfected endothelial cells alone cannot induce CD1d [53], indicating that a separate signal besides cytokine exposure is necessary for de novo CD1d synthesis and that this signal must be generated through direct virus-cell interactions. In fact, CVB3 binding to decay accelerating factor (DAF) one of the two know cellular receptors for [54], provides the essential secondary signal [17]. DAF is a glycosylphosphatidylinositol (GPI)-anchored membrane glycoprotein and its primary biological function is to prevent autologous complement induced cell injury by inactivating C3 [55–57]. Signaling through this molecule by C3 or CVB3 induces calcium flux and activation of the transcription factor, NFAT [58, 59]. CVB3 infection of DAF deficient cells fails to induce CD1d expression [17], and blocking NFAT activation either by use of cells expressing a dominant-negative-NFAT or by cyclosporine A prevents CD1d expression in virus infected cells. Thus, only cells which are exposed to TNFα and bind virus to DAF on the cell membrane will up-regulate CD1d.
While many microbial infections augment CD1 expression, other infectious agents suppress expression of these molecules on dendritic cells and antigen presenting cells [60–62]. In Leishmania, cytomegalovirus, and herpes simplex virus infections, CD1 expression is down-regulated. With HSV, modulation of CD1 was dependent upon the amount of virus with low levels of virus enhancing both type 1 and 2 CD1 expression on human dendritic cells while high levels of virus suppressed CD1 expression. In HSV, suppression correlated with accumulation of intracellular viral protein and interruption of CD1 recycling pathway. However, other studies demonstrate that activating TLR7/8, TLR recognizing single strand RNA and RNA viruses, block CD1 expression at the protein and mRNA levels [63]. Where CD1-restricted immunity promotes host defense against infection, inhibition of CD1 up-regulation can provide an evasion mechanism for the microbes.
Role of CD1d in Innate Immunity and Infection
CD1d-dependent innate immunity is important in a wide range of diseases. Infections with P. aeruginosa[64], C. neoformans[65], Herpes Simplex virus [66], Hepatitis C virus [50], and encephalomyocarditis virus [67, 68] make the disease substantially worse in animals lacking CD1 or NKT cells. In contrast, clearance of RSV is delayed in CD1d deficient mice [49, 69]. CD1-restricted cells appear to have minimal effects in cytomegalovirus and lymphocytic choriomeningitis virus infections [70]. In Novosphingobium aromaticivorans infection in mice, CD1d presentation of alpha-glycuronosylceramide from the bacterial cell wall activates NKT cells and ultimately results in liver-specific autoimmunity [37]. This means that CD1d-restricted effectors may either have no, beneficial or detrimental functions depending upon the infectious agent.
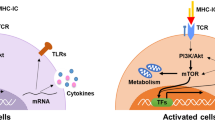
There are two major populations of CD1-restricted T cells. These are NKT cells and γδ T cells. NKT cells co-express T cell receptors (TCR) and NK receptors. There are two types of NKT cells. Type 1 NKT cells have a TCR comprised of a single type of TCRα chain (Vα14Jα18 for mice and Vα24Jα18 for humans) and one of a limited number of distinct TCRβ chains resulting in limited clonal diversity. These cells are usually designated as invariant NKT (iNKT) cells. Type 2 NKT cells use diverse TCR (non-Vα14Jα18/Vα24Jα18). Both Type 1 and Type2 NKT cells are CD1d restricted [71–73]. NKT cells comprise up to 2% of spleen, 20% of mononuclear cells in the liver and 40% of CD3+ cells in bone marrow in the mouse making these cells a major component of the total T cell population [74, 75]. NKT cells have a constitutively activated phenotype and are capable of rapidly secreting large amounts of cytokines (IFN-γ, IL-4, IL-17, IL-5, and IL-13) upon activation, which can modulate many immunological processes, including tumor immunity, maintenance of immunologic self tolerance, prevention of autoimmune disorders, and protection from a variety of pathogens during experimental infections [73, 76, 77]. Rapid cytokine secretion occurs because cytokine mRNAs pre-exist in the cells [78]. The presence of pro-inflammatory cytokines (IL-12 and IL-12/IL-18) can dramatically reduce the amount of CD1-dependent TCR signaling required for NKT cell activation [79, 80]. Type 1 NKT cells produce IFNγ and IL-2 which activates NK cells and dendritic cells, enhancing antigen presentation [81, 82]. Rapid cytokine secretion by the NKT cells polarizes developing adaptive immunity along the Th1/Th2 axis [83]. Both Type 1 and Type 2 NKT cells can have a Th1 or Th2 phenotype with corresponding cytokine profiles, and therefore may have either potentiating or protective roles in infections and autoimmune diseases [84]. The majority of reports indicate that Type 2 NKT cells are protective in autoimmune diseases in mice including in models of autoimmune diabetes in NOD mice [85], EAE [86] and Con-A induced hepatitis [87]. Furthermore, while type 1 NKT cells may increase tumor immunosurveillance, type 2 NKT cells may suppress anti-tumor immunity [74, 88].
NKT cells are not the only CD1 restricted lymphocyte. Human γδ T cells recognize lipid antigens presented by CD1 [89, 90], and mouse γδ T cells recognize non-classical MHC antigens (T10/T22) [91, 92]. γδ cells expressing the Vγ4 TCR recognize CD1d [16]. Within 2-4 days of CVB3 infection, CD1d is rapidly up-regulated on cardiac endothelial cells and myocytes and results in infiltration of Vγ4+ T cells into the myocardium [93]. As with NKT cells, Vγ4+ cells rapidly secrete large amounts of pro-inflammatory cytokines including TNFα and IFNγ which establish an environment conducive to polarizing the developing virus specific adaptive immune response to a Th1 phenotype [94, 95]. The Vγ4+ cells also kill CVB3 infected CD1d+ cardiocytes in a Fas-dependent manner which aids in viral control by eliminating infected cells early in the virus replication cycle. As with NKT cells, γδ cells can interact with CD1 on dendritic cells/macrophage resulting in enhanced antigen presentation and cytokine release [96, 97]. CVB3 infection of mice lacking γδ T cells results in increased virus titers in the heart but little or no cardiac inflammation, animal mortality, or heart-specific autoimmunity[98–100] making γδ cells essential in the pathogenesis of CVB3 infections.
Several cases of clinical cardiomyopathy where γδ cells dominate the inflammatory infiltrate have been reported [101–103] suggesting that these innate effectors can be directly pathogenic. More often, γδ cells would impact myocarditis through their effects on the antigen-specific, adaptive immune response. In this laboratory's mouse model of CVB3 induced myocarditis, infection activates heart-specific, autoimmune CD8+ cytolytic T lymphocytes [12, 100, 104, 105] which kill uninfected cardiocytes through recognition of cardiac myosin epitopes [14] and can adoptively transfer myocarditis into uninfected recipients [106]. These autoimmune CD8+ effectors are the primary cause of cardiac injury. In vivo generation of autoimmune CD8+ cells requires activated Vγ4 cells and mice lacking either the Vγ4 or all γδ cells do not generate autoimmunity [100]. Thus, the primary role of Vγ4 cells in CVB3 induced myocarditis is to facilitate autoimmunity induction.
Role of CD1d in CD4+CD25+ regulatory T cell response
Regulatory T cells (Tregs) are important negative immune modulators, constitute up to 10% of peripheral CD4+ T cells in naive mice and humans, and express CD25 (IL-2 receptor α chain) [107–109]. Several subsets of T regulatory cells have been described and these can basically be divided into natural (nTreg) and inducible (iTreg) populations. The nTreg cells arise in the thymus during normal T cell ontogeny as CD4+CD25+ cells and depend upon expression of the FoxP3 transcription factor. Indeed FoxP3 expression is crucial to the immunosuppression activity of these nTreg since transduction of exogenous FoxP3 into CD4+CD25- cells is sufficient to convert these cells into CD4+CD25+ Treg cells [108]. Developing T cells with high affinity TCR for self antigens are most probably committed to the nTreg line. While most αβ TCR+ cells (exclusive of NKT cells) developing in the thymus enter the periphery as naïve cells, nTreg cells are functionally mature when leaving the thymus and do not require antigen exposure peripherally to generate immunosuppressive activity. While FoxP3 is necessary for conversion of CD4+ cells to Treg cells, IL-2 is required for Treg cell maintenance/survival. Animals lacking either CD25 (IL-2R) or IL-2 develop lymphoproliferative and autoimmune diseases [110] associated with a decrease in Treg cells. Although the transcription factor NFAT normally increases expression of IL-2and IFNγ while decreasing expression of CD25 and CTLA4, NFAT complexed with FoxP3 has the opposite effect, decreasing IL-2/IFNγ and increasing CD25/CTLA4 expression. In addition to nTreg cells, inducible regulatory T cells (iTreg) can be converted from effector T cell populations in the periphery subsequent to antigen challenge. These iTreg cells are CD4+CD25+ but can either be FoxP3+ or FoxP3- [111]. Both iTreg and nTreg cells can secret either IL-10 (Tr1) or TGFβ (Th3) cytokines [111]. Treg cells express similar chemokine receptor patterns as effector T cells and can migrate to peripheral lymphoid tissues and inflammatory sites similarly to the effector population [108]. There are three general hypothesized mechanisms for Treg cell suppression of immunity [108]. First, T reg cells may bind to and out-compete effector T cells for MHC-antigen complexes on dendritic cells and effectively block antigen presentation to the effector T cell populations. Secondly, direct Treg-dendritic cell interactions through CTLA4 can down-regulate accessory molecule expression (CD80/CD86) on the dendritic cells making them less effective in antigen presentation. Third, Treg cells can either kill or inhibit T cell differentiation. TGFβ produced by Treg cells will activate NOTCH and its downstream target gene, Hes1, which suppresses gene expression in T cells [111]. IL-10 blocks CD2, CD28 and ICOS signaling in T cells and SOCS3 signaling in monocytes resulting in reduced T cell proliferation and cytokine response [112].
Treg cells play an important role in preventing autoimmunity in myocarditis [19, 113, 114]. Two CVB3 variants have been identified which differ by a single non-conserved mutation in the VP2 capsid protein in a region associated with DAF binding [15]. One variant, designated H3, binds with high avidity to DAF, causes calcium flux and NFAT activation, induces CD1d expression in the heart and activates Vγ4 cells [16, 17, 19, 59]. The other variant, designated H310A1, binds with low affinity to DAF, fails to activate NFAT, does not up-regulate CD1d expression in the heart, and does not activate Vγ4 cells. While H3 virus induces autoimmune CD8 T cells and causes severe myocarditis, the H310A1 virus fails to induce autoimmunity and induces minimal cardiac injury despite high virus titers in the heart [115]. The primary difference between the two virus infections is that H310A1 infection activates CD4+CD25+FoxP3+ Treg cells which are absent in H3 infected mice [19].
Innate effector T cells control Treg cell responses. Although somewhat controversial, various reports indicate that iNKT cells suppress autoimmunity by promoting T regulatory cell activation. Studies investigating oral tolerance to nickel demonstrated that antigen presenting cells interact with type 1 NKT cells through CD1d causing the NKT cells to secrete IL-4 and IL-10 and activate Treg cells [116–118]. Similar studies found that T regulatory cells fail to generate in CD1dKO mice [119] and iNKT KO mice [119]. Other studies show that αGalCer, a well known and specific NKT CD1d-restricted ligand, increases Treg cell numbers in vivo [120] and can suppress autoimmune diabetes in NOD mice [121–123]. NKT cells secret high levels of TGFβ and IL-10 [124, 125] which alter dendritic cell (DC) cytokine (IL-10) and accessory molecule (CD40, CD80 and/or CD86) expression [126–128] that favors T regulatory cell responses [129, 130].
A number of reports indicate that γδ T cells can also affect Treg cell responses. IL-23 activated γδ cells prevent conversion of effector T cells to iTreg cells [131]. Similarly γδ cells reduce IL-10 producing Treg cells in the lung in an asthma model γδ cells [132]. Vγ2Vδ2 cells prevent IL-2 induced expansion of CD4+CD25+FoxP3+ T [133]. Therefore, while many studies suggest that NKT cells promote Treg cell activation and protect from autoimmunity, it appears that γδ T cells can have the opposite effect and promote autoimmunity/inflammation through inhibiting T regulatory cell activity. As with NKT cells, the mechanism by which γδ T cells modulate Treg cell responses can be diverse and include alterations in antigen presenting cells which prevent Treg cell activation, and suppression of IL2 needed for Treg cell maintenance. This laboratory has recently reported an additional mechanism for γδ T cell modulation of Treg cell responses using two coxsackievirus B3 variants which differ by a single non-conserved amino acid in the VP2 capsid protein [15]. These studies show that the non-pathogenic variant induces a potent T regulatory cell (CD4+CD25+FoxP3+) response which is absent during infections with the pathogenic virus [19]. Although H3 infected mice normally have few CD4+CD25+FoxP3+ cells, H3 infection of γδ cell deficient mice results in significant increases in Treg cells and suppression of myocarditis. Therefore, this study, as others mentioned above, find that γδ cells antagonize Treg cell responses and promote autoimmunity. Further studies demonstrate that a subpopulation of Treg cells in γδ deficient mice express high levels of CD1d, that the CD1d+ Treg cells are substantially more immunosuppressive on a per cell basis than CD1d- Treg cells, and that Vγ4+ cells selectively kill the CD1d+ Treg cells in a CD1d and caspase-dependent manner [10].
Conclusions
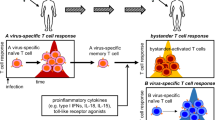
Innate immunity is crucial to anti-microbial host defense as it helps control infectious agents until a more potent microbe-specific, adaptive immune response can be generated. However, the innate response is also important in molding the nature of the adaptive immune response. CD1 molecules, as members of the non-classical MHC family, are intimately involved in innate immunity. NKT and a subset of γδ T cells are CD1 restricted. Figure 1 provides a schematic of the interactions between innate effectors and the adaptive immune response through CD1d. This raises the interesting question of why two innate effector populations would exist which respond to the same type of antigen presenting molecule, especially since CD1 molecules have limited diversity and are therefore more likely to present less heterogeneous antigens than classical MHC antigens. Two possibilities are that CD1-restricted NKT and γδ T cells have redundant functions or that each type of CD1-restricted effector has a distinct role in the immune response. One potential reason for redundancy is that NKT and γδ cells tend to concentrate in different tissues. NKT cells comprise 20-30% of liver and bone marrow T cells but are generally absent in intestinal epithelial lymphocytes (IEL) [134]. In contrast, γδ concentrate in epithelia of skin, intestine and reproductive tract where these cells can comprise up to 50% of the T cells [135]. This distribution could imply that CD1 presentation of microbial or self antigens will preferentially activate NKT or γδ cells depending upon the tissue involved. In peripheral lymphoid organs where both NKT and γδ cells are present, other factors must determine if CD1-dependent NKT or γδ cell responses predominate. What these factors are is not known. One possibility is that while CD1 presents a variety of glycolipid antigens, NKT and γδ cells recognize distinct sets of these antigens. In this case, CD1d might exclusively activate only NKT or γδ cells depending upon which glycolipid antigens are presented. A second possibility is that the binding avidity for CD1d-antigen complexes differ for NKT and γδ cells meaning that the innate effector with the stronger binding avidity would dominate. The relevant point is that the balance between NKT and γδ activation can be the deciding factor between self-tolerance and autoimmunity. The major unresolved question is what decides whether γδ or NKT cells dominate in an innate response where CD1 is up-regulated.

CVB3 up-regulates CD1d on dendritic cells and non-hemopoietic cells such as cardiac myocytes. CD1d-glycolipid complexes on dendritic cells activate NKT and Vγ4+ T cells which kill virus infected cells limit virus infections, while the activated NKT/Vγ4+ cells also alter dendritic cells. CTLR4 expression by NKT cells either blocks or down-regulates B7 co-stimulatory molecules inhibiting antigen presentation to antigen-specific CD4+ and CD8+ T cells, and promoting inducible Tregulatory cell activation through release of TGFβ, IL-10 and IL-2. TNFα and IFNγ secretion by Vγ4+ T cells enhances maturation and antigen presentation by dendritic cells but suppresses activation of the inducible Tregulatory cell. CVB3 infection up-regulates CD1d expression on a subpopulation of CD4+CD25+FoxP3+ Tregulatory cells. The CD1d+Tregulatory cells are substantially more immunosuppressive than CD1d- Tregulatory cells and are primarily responsible for preventing autoimmunity to cardiac antigens and myocarditis during CVB3 infection. Vγ4+ T cells selectively kill CD1d+ T regulatory cells through caspase-dependent apoptosis which then results in autoimmunity induction. Thus, pathogenesis in CVB3 infection depends upon the balance in NKT and Vγ4+ T cell activation.
Conflict of interest statement
The author states that they have no conflict of interest.
References
Liu WLW, Gao C, Sun NL: Effects of atorvastatin on the Th1/Th2 polarization of ongoing experimental autoimmune myocarditis in Lewis rats. J Autoimmun 2005, 25: 258-263. 10.1016/j.jaut.2005.06.005
Woodruff J: Viral myocarditis. Am J Pathol 1980, 101: 425-483.
Fabre ASM: Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death: a UK experience. Heart 2006, 92: 316-320. 10.1136/hrt.2004.045518
Doolan ALN, Semsarian C: Causes of sudden cardiac death in young Australians. Med J Aust 2004, 180: 110-112.
Eckart RE, Scoville SL, Campbell CL, Shry EA, Stajduhar KC, Potter RN, Pearse LA, Virmani R: Sudden death in young adults: a 25-year review of autopsies in military recruits. Ann Intern Med 2004, 141: 829-834.
Solberg EE, Gjertsen F, Haugstad E, Kolsrud L: Sudden death in sports among young adults in Norway. Eur J Cardiovasc Prev Rehabil 2010, 17: 337-341.
Passarino G, Burlo P, Ciccone G, Comino A, Cravello M, Iannicelli P, Mollo F: Prevalence of myocarditis at autopsy in Turin, Italy. Arch Pathol Lab Med 1997, 121: 619-622.
Friman G, Wesslen L, Fohlman J, Karjalainen J, Rolf C: The epidemiology of infectious myocarditis, lymphocytic and dilated cardiomyopathy. Eur Heart J 1995, 16: 36-41.
Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R, Hare J, Bricker JT, Bowles KR, Towbin JA: Detection of viruses in myocardial tissues by polymerase chain reaction. evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol 2003, 42: 466-472. 10.1016/S0735-1097(03)00648-X
Huber S: γδ T lymphocytes kill T regulatory cells through CD1d. Immunology 2010, 131: 202-209. 10.1111/j.1365-2567.2010.03292.x
Woodruff J, Woodruff J: Involvement of T lymphocytes in the pathogenesis of coxsackievirus B3 heart disease. J Immunol 1974, 113: 1726-1734.
Guthrie M, Lodge PA, Huber SA: Cardiac injury in myocarditis induced by Coxsackievirus group B, type 3 in Balb/c mice is mediated by Lyt 2 + cytolytic lymphocytes. Cell Immunol 1984, 88: 558-567. 10.1016/0008-8749(84)90188-6
Huber S, Cunningham M: Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsackiebiral myocarditis. J Immunol 1996, 156: 3528-3534.
Huber S, Gauntt C: Antigenic mimicry between self and coxsackievirus protein leads to both humoral and cellular autoimmunity to heart proteins. In Molecular Mimicry, Microbes, and Immunity. Edited by: Cunningham M, Fujinami R. Washington DC: ASM Press; 2000:57-68.
Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber S: A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol 1996, 70: 7811-7818.
Huber S, Sartini D, Exley M: Role of CD1d in coxsackievirus B3-induced myocarditis. J Immunol 2003, 170: 3147-3153.
Huber S, Song WC, Sartini D: Decay-accelerating factor (CD55) promotes CD1d expression and Vgamma4+ T-cell activation in coxsackievirus B3-induced myocarditis. Viral Immunol 2006, 19: 156-166. 10.1089/vim.2006.19.156
Huber SA: CD1d expression on hemopoietic cells promotes CD4+ Th1 response in coxsackievirus B3 induced myocarditis. Virology 2006, 352: 226-236. 10.1016/j.virol.2006.04.014
Huber SA, Feldman AM, Sartini D: Coxsackievirus B3 induces T regulatory cells, which inhibit cardiomyopathy in tumor necrosis factor-alpha transgenic mice. Circ Res 2006, 99: 1109-1116. 10.1161/01.RES.0000249405.13536.49
Kulkarni RRHS, Sharif S: The invariant NKT cell subset in anti-viral defenses: a dark horse in anti-influenza immunity? J Leukoc Biol 2010, 88: 635-643. 10.1189/jlb.0410191
Calabi F, Jarvis JM, Martin L, Milstein C: Two classes of CD1 genes. Eur J Immunol 1989, 19: 285-292. 10.1002/eji.1830190211
Blumberg RS, Gerdes D, Chott A, Porcelli SA, Balk SP: Structure and function of the CD1 family of MHC-like cell surface proteins. Immunol Rev 1995, 147: 5-29. 10.1111/j.1600-065X.1995.tb00085.x
de la Salle H, Mariotti S, Angenieux C, Gilleron M, Garcia-Alles LF, Malm D, Berg T, Paoletti S, Maitre B, Mourey L, et al.: Assistance of microbial glycolipid antigen processing by CD1e. Science 2005, 310: 1321-1324. 10.1126/science.1115301
Exley M, Garcia J, Wilson SB, Spada F, Gerdes D, Tahir SM, Patton KT, Blumberg RS, Porcelli S, Chott A, Balk SP: CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology 2000, 100: 37-47. 10.1046/j.1365-2567.2000.00001.x
Boes M, Stoppelenburg AJ, Sille FC: Endosomal processing for antigen presentation mediated by CD1 and Class I major histocompatibility complex: roads to display or destruction. Immunology 2009, 127: 163-170. 10.1111/j.1365-2567.2009.03078.x
Odyniec AN, Barral DC, Garg S, Tatituri RV, Besra GS, Brenner MB: Regulation of CD1 antigen-presenting complex stability. J Biol Chem 2010, 285: 11937-11947. 10.1074/jbc.M109.077933
Brutkiewicz RR, Bennink JR, Yewdell JW, Bendelac A: TAP-independent, beta 2-microglobulin-dependent surface expression of functional mouse CD1.1. J Exp Med 1995, 182: 1913-1919. 10.1084/jem.182.6.1913
Cheng TY, Relloso M, Van Rhijn I, Young DC, Besra GS, Briken V, Zajonc DM, Wilson IA, Porcelli S, Moody DB: Role of lipid trimming and CD1 groove size in cellular antigen presentation. EMBO J 2006, 25: 2989-2999. 10.1038/sj.emboj.7601185
Zajonc DM, Elsliger MA, Teyton L, Wilson IA: Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 A. Nat Immunol 2003, 4: 808-815. 10.1038/ni948
Batuwangala T, Shepherd D, Gadola SD, Gibson KJ, Zaccai NR, Fersht AR, Besra GS, Cerundolo V, Jones EY: The crystal structure of human CD1b with a bound bacterial glycolipid. J Immunol 2004, 172: 2382-2388.
Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB: Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature 1994, 372: 691-694. 10.1038/372691a0
Sieling PA, Chatterjee D, Porcelli SA, Prigozy TI, Mazzaccaro RJ, Soriano T, Bloom BR, Brenner MB, Kronenberg M, Brennan PJ, et al.: CD1-restricted T cell recognition of microbial lipoglycan antigens. Science 1995, 269: 227-230. 10.1126/science.7542404
Moody DB: Polyisoprenyl glycolipids as targets of CD1-mediated T cell responses. Cell Mol Life Sci 2001, 58: 1461-1474. 10.1007/PL00000789
Shamshiev A, Donda A, Prigozy TI, Mori L, Chigorno V, Benedict CA, Kappos L, Sonnino S, Kronenberg M, De Libero G: The alphabeta T cell response to self-glycolipids shows a novel mechanism of CD1b loading and a requirement for complex oligosaccharides. Immunity 2000, 13: 255-264. 10.1016/S1074-7613(00)00025-X
Beckman EM, Melian A, Behar SM, Sieling PA, Chatterjee D, Furlong ST, Matsumoto R, Rosat JP, Modlin RL, Porcelli SA: CD1c restricts responses of mycobacteria-specific T cells. Evidence for antigen presentation by a second member of the human CD1 family. J Immunol 1996, 157: 2795-2803.
Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M: Recognition of bacterial glycosphingolipids by natural killer T cells. Nature 2005, 434: 520-525. 10.1038/nature03407
Mattner J, Savage PB, Leung P, Oertelt SS, Wang V, Trivedi O, Scanlon ST, Pendem K, Teyton L, Hart J, et al.: Liver autoimmunity triggered by microbial activation of natural killer T cells. Cell Host Microbe 2008, 3: 304-315. 10.1016/j.chom.2008.03.009
Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, Zajonc DM, Ben-Menachem G, Ainge GD, Painter GF, et al.: Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol 2006, 7: 978-986. 10.1038/ni1380
Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, et al.: Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 2005, 434: 525-529. 10.1038/nature03408
Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, et al.: CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997, 278: 1626-1629. 10.1126/science.278.5343.1626
Roy KC, Maricic I, Khurana A, Smith TR, Halder RC, Kumar V: Involvement of secretory and endosomal compartments in presentation of an exogenous self-glycolipid to type II NKT cells. J Immunol 2008, 180: 2942-2950.
Franchini L, Matto P, Ronchetti F, Panza L, Barbieri L, Costantino V, Mangoni A, Cavallari M, Mori L, De Libero G: Synthesis and evaluation of human T cell stimulating activity of an alpha-sulfatide analogue. Bioorg Med Chem 2007, 15: 5529-5536. 10.1016/j.bmc.2007.05.044
Hegde S, Fox L, Wang X, Gumperz JE: Autoreactive natural killer T cells: promoting immune protection and immune tolerance through varied interactions with myeloid antigen-presenting cells. Immunology 2010, 130: 471-483. 10.1111/j.1365-2567.2010.03293.x
Darmoise A, Teneberg S, Bouzonville L, Brady RO, Beck M, Kaufmann SH, Winau F: Lysosomal alpha-galactosidase controls the generation of self lipid antigens for natural killer T cells. Immunity 2010, 33: 216-228. 10.1016/j.immuni.2010.08.003
Roura-Mir C, Wang L, Cheng TY, Matsunaga I, Dascher CC, Peng SL, Fenton MJ, Kirschning C, Moody DB: Mycobacterium tuberculosis regulates CD1 antigen presentation pathways through TLR-2. J Immunol 2005, 175: 1758-1766.
Moody DB: The surprising diversity of lipid antigens for CD1-restricted T cells. Adv Immunol 2006, 89: 87-139. full_text
Dougan SK, Kaser A, Blumberg RS: CD1 expression on antigen-presenting cells. Curr Top Microbiol Immunol 2007, 314: 113-141. full_text
Sallusto F, Lanzavecchia A: Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 1994, 179: 1109-1118. 10.1084/jem.179.4.1109
Skold M, Behar SM: Role of CD1d-restricted NKT cells in microbial immunity. Infect Immun 2003, 71: 5447-5455. 10.1128/IAI.71.10.5447-5455.2003
Durante-Mangoni E, Wang R, Shaulov A, He Q, Nasser I, Afdhal N, Koziel MJ, Exley MA: Hepatic CD1d expression in hepatitis C virus infection and recognition by resident proinflammatory CD1d-reactive T cells. J Immunol 2004, 173: 2159-2166.
Skold M, Xiong X, Illarionov PA, Besra GS, Behar SM: Interplay of cytokines and microbial signals in regulation of CD1d expression and NKT cell activation. J Immunol 2005, 175: 3584-3593.
Raghuraman G, Geng Y, Wang CR: IFN-beta-mediated up-regulation of CD1d in bacteria-infected APCs. J Immunol 2006, 177: 7841-7848.
Huber SA, Sartini D: Roles of tumor necrosis factor alpha (TNF-alpha) and the p55 TNF receptor in CD1d induction and coxsackievirus B3-induced myocarditis. J Virol 2005, 79: 2659-2665. 10.1128/JVI.79.5.2659-2665.2005
Bergelson JM, Mohanty JG, Crowell RL, St John NF, Lublin DM, Finberg RW: Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J Virol 1995, 69: 1903-1906.
Song WC, Deng C, Raszmann K, Moore R, Newbold R, McLachlan JA, Negishi M: Mouse decay-accelerating factor: selective and tissue-specific induction by estrogen of the gene encoding the glycosylphosphatidylinositol-anchored form. J Immunol 1996, 157: 4166-4172.
Miwa T, Maldonado MA, Zhou L, Sun X, Luo HY, Cai D, Werth VP, Madaio MP, Eisenberg RA, Song WC: Deletion of decay-accelerating factor (CD55) exacerbates autoimmune disease development in MRL/lpr mice. Am J Pathol 2002, 161: 1077-1086. 10.1016/S0002-9440(10)64268-X
Miwa T, Sun X, Ohta R, Okada N, Harris CL, Morgan BP, Song WC: Characterization of glycosylphosphatidylinositol-anchored decay accelerating factor (GPI-DAF) and transmembrane DAF gene expression in wild-type and GPI-DAF gene knockout mice using polyclonal and monoclonal antibodies with dual or single specificity. Immunology 2001, 104: 207-214. 10.1046/j.1365-2567.2001.01280.x
Peiffer I, Servin AL, Bernet-Camard MF: Piracy of decay-accelerating factor (CD55) signal transduction by the diffusely adhering strain Escherichia coli C1845 promotes cytoskeletal F-actin rearrangements in cultured human intestinal INT407 cells. Infect Immun 1998, 66: 4036-4042.
Huber SA, Rincon M: Coxsackievirus B3 induction of NFAT: requirement for myocarditis susceptibility. Virology 2008, 381: 155-160. 10.1016/j.virol.2008.08.020
Raftery MJ, Hitzler M, Winau F, Giese T, Plachter B, Kaufmann SH, Schonrich G: Inhibition of CD1 antigen presentation by human cytomegalovirus. J Virol 2008, 82: 4308-4319. 10.1128/JVI.01447-07
Donovan MJ, Jayakumar A, McDowell MA: Inhibition of groups 1 and 2 CD1 molecules on human dendritic cells by Leishmania species. Parasite Immunol 2007, 29: 515-524. 10.1111/j.1365-3024.2007.00970.x
Raftery MJ, Winau F, Kaufmann SH, Schaible UE, Schonrich G: CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J Immunol 2006, 177: 6207-6214.
Assier E, Marin-Esteban V, Haziot A, Maggi E, Charron D, Mooney N: TLR7/8 agonists impair monocyte-derived dendritic cell differentiation and maturation. J Leukoc Biol 2007, 81: 221-228. 10.1189/jlb.0705385
Hazlett LD, Li Q, Liu J, McClellan S, Du W, Barrett RP: NKT Cells Are Critical to Initiate an Inflammatory Response after Pseudomonas aeruginosa Ocular Infection in Susceptible Mice. J Immunol 2007, 179: 1138-1146.
Kawakami K, Kinjo Y, Yara S, Koguchi Y, Uezu K, Nakayama T, Taniguchi M, Saito A: Activation of Valpha14(+) natural killer T cells by alpha-galactosylceramide results in development of Th1 response and local host resistance in mice infected with Cryptococcus neoformans. Infect Immun 2001, 69: 213-220. 10.1128/IAI.69.1.213-220.2001
Grubor-Bauk B, Arthur JL, Mayrhofer G: Importance of NKT cells in resistance to herpes simplex virus, fate of virus-infected neurons, and level of latency in mice. J Virol 2008, 82: 11073-11083. 10.1128/JVI.00205-08
Ilyinskii PO, Wang R, Balk SP, Exley MA: CD1d mediates T-cell-dependent resistance to secondary infection with encephalomyocarditis virus (EMCV) in vitro and immune response to EMCV infection in vivo. J Virol 2006, 80: 7146-7158. 10.1128/JVI.02745-05
Exley MA, Bigley NJ, Cheng O, Shaulov A, Tahir SM, Carter QL, Garcia J, Wang C, Patten K, Stills HF, et al.: Innate immune response to encephalomyocarditis virus infection mediated by CD1d. Immunology 2003, 110: 519-526. 10.1111/j.1365-2567.2003.01779.x
Johnson TR, Hong S, Van Kaer L, Koezuka Y, Graham BS: NK T cells contribute to expansion of CD8(+) T cells and amplification of antiviral immune responses to respiratory syncytial virus. J Virol 2002, 76: 4294-4303. 10.1128/JVI.76.9.4294-4303.2002
Harker JAGA, Wahlsten JL, Lee DC, Thorne LG, Sawant D, Tregoning JS, Caspi RR, Bukreyev A, Collins PL, Openshaw PJ: Interleukin 18 coexpression during respiratory syncytial virus infection results in enhanced disease mediated by natural killer cells. J Virol 2010, 84: 4073-4082. 10.1128/JVI.02014-09
Taniguchi M, Tashiro T, Dashtsoodol N, Hongo N, Watarai H: The specialized iNKT cell system recognizes glycolipid antigens and bridges the innate and acquired immune systems with potential applications for cancer therapy. Int Immunol 2010, 22: 1-6. 10.1093/intimm/dxp104
Ronchi F, Falcone M: Immune regulation by invariant NKT cells in autoimmunity. Front Biosci 2008, 13: 4827-4837. 10.2741/3042
Kronenberg M: Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol 2005, 3: 877-900. 10.1146/annurev.immunol.23.021704.115742
Terabe M, Berzofsky JA: The role of NKT cells in tumor immunity. Adv Cancer Res 2008, 101: 277-348. 10.1016/S0065-230X(08)00408-9
Bendelac A, Savage PB, Teyton L: The biology of NKT cells. Annu Rev Immunol 2007, 25: 297-336. 10.1146/annurev.immunol.25.022106.141711
Olson CM Jr, Bates TC, Izadi H, Radolf JD, Huber SA, Boyson JE, Anguita J: Local production of IFN-gamma by invariant NKT cells modulates acute Lyme carditis. J Immunol 2009, 182: 3728-3734. 10.4049/jimmunol.0804111
Michel MLKA, Paget C, Fujio M, Trottein F, Savage PB, Wong CH, Schneider E, Dy M, Leite-de-Moraes MC: Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med 2007, 204: 995-1001. 10.1084/jem.20061551
Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, Kronenberg M, Locksley RM: Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med 2003, 198: 1069-1076. 10.1084/jem.20030630
Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB: Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol 2003, 4: 1230-1237. 10.1038/ni1002
Seki S, Habu Y, Kawamura T, Takeda K, Dobashi H, Ohkawa T, Hiraide H: The liver as a crucial organ in the first line of host defense: the roles of Kupffer cells, natural killer (NK) cells and NK1.1 Ag+ T cells in T helper 1 immune responses. Immunol Rev 2000, 174: 35-46. 10.1034/j.1600-0528.2002.017404.x
Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, Bendelac A: Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol 1999, 163: 4647-4650.
Gigli G, Caielli S, Cutuli D, Falcone M: Innate immunity modulates autoimmunity: type 1 interferon-beta treatment in multiple sclerosis promotes growth and function of regulatory invariant natural killer T cells through dendritic cell maturation. Immunology 2007, 122: 409-417. 10.1111/j.1365-2567.2007.02655.x
Wiethe C, Debus A, Mohrs M, Steinkasserer A, Lutz M, Gessner A: Dendritic cell differentiation state and their interaction with NKT cells determine Th1/Th2 differentiation in the murine model of Leishmania major infection. J Immunol 2008, 180: 4371-4381.
Arrenberg P, Halder R, Kumar V: Cross-regulation between distinct natural killer T cell subsets influences immune response to self and foreign antigens. J Cell Physiol 2009, 218: 246-250. 10.1002/jcp.21597
Duarte N, Stenstrom M, Campino S, Bergman ML, Lundholm M, Holmberg D, Cardell SL: Prevention of diabetes in nonobese diabetic mice mediated by CD1d-restricted nonclassical NKT cells. J Immunol 2004, 173: 3112-3118.
Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V: Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med 2004, 199: 947-957. 10.1084/jem.20031389
Halder RC, Aguilera C, Maricic I, Kumar V: Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest 2007, 117: 2302-2312. 10.1172/JCI31602
Ambrosino E, Terabe M, Halder RC, Peng J, Takaku S, Miyake S, Yamamura T, Kumar V, Berzofsky JA: Cross-regulation between type I and type II NKT cells in regulating tumor immunity: a new immunoregulatory axis. J Immunol 2007, 179: 5126-5136.
Spada FM, Grant EP, Peters PJ, Sugita M, Melian A, Leslie DS, Lee HK, van Donselaar E, Hanson DA, Krensky AM, et al.: Self-recognition of CD1 by gamma/delta T cells: implications for innate immunity. J Exp Med 2000, 191: 937-948. 10.1084/jem.191.6.937
Cui Y, Kang L, Cui L, He W: Human gammadelta T cell recognition of lipid A is predominately presented by CD1b or CD1c on dendritic cells. Biol Direct 2009, 4: 47. 10.1186/1745-6150-4-47
Van Kaer L, Wu M, Ichikawa Y, Ito K, Bonneville M, Ostrand-Rosenberg S, Murphy DB, Tonegawa S: Recognition of MHC TL gene products by gamma delta T cells. Immunol Rev 1991, 120: 89-115. 10.1111/j.1600-065X.1991.tb00589.x
Chien YH, Konigshofer Y: Antigen recognition by gammadelta T cells. Immunol Rev 2007, 215: 46-58. 10.1111/j.1600-065X.2006.00470.x
Huber SA: Increased susceptibility of male BALB/c mice to coxsackievirus B3-induced myocarditis: role for CD1d. Med Microbiol Immunol 2005, 194: 121-127. 10.1007/s00430-004-0221-6
Huber S, Mortensen A, Moulton G: Modulation of cytokine expression by CD4+ T cells during coxsackievirus B3 infections of BALB/c mice initiated by cells expressing the gamma-delta+ T cell receptor. J Virol 1996, 70: 3039-3045.
Huber S, Graveline D, Born W, O'Brien R: Cytokine Production by Vgamma+ T Cell Subsets is an Important Factor Determining CD4+ Th Cell Phenotype and Susceptibility of BALB/c mice to Coxsackievirus B3-Induced Myocarditis. J Virol 2001, 75: 5860-5868. 10.1128/JVI.75.13.5860-5869.2001
Leslie DS, Vincent MS, Spada FM, Das H, Sugita M, Morita CT, Brenner MB: CD1-mediated gamma/delta T cell maturation of dendritic cells. J Exp Med 2002, 196: 1575-1584. 10.1084/jem.20021515
Collins C, Wolfe J, Roessner K, Shi C, Sigal LH, Budd RC: Lyme arthritis synovial gammadelta T cells instruct dendritic cells via fas ligand. J Immunol 2005, 175: 5656-5665.
Huber S, Moraska A, Choate M: T cells expressing the gamma delta T-cell receptor potentiate coxsackievirus B3-induced myocarditis. J Virol 1992, 66: 6541-6546.
Huber SA: Depletion of gammadelta+ T cells increases CD4+ FoxP3 (T regulatory) cell response in coxsackievirus B3-induced myocarditis. Immunology 2009, 127: 567-576. 10.1111/j.1365-2567.2008.03034.x
Huber SA, Sartini D, Exley M: Vgamma4(+) T cells promote autoimmune CD8(+) cytolytic T-lymphocyte activation in coxsackievirus B3-induced myocarditis in mice: role for CD4(+) Th1 cells. J Virol 2002, 76: 10785-10790. 10.1128/JVI.76.21.10785-10790.2002
Takeda N, Seko Y, Oriuchi N, Nagai R: Gamma-delta T-cell-mediated dilated cardiomyopathy. Int J Cardiol 2008, 125: 130-132. 10.1016/j.ijcard.2007.01.068
Takeda N, Takahashi T, Seko Y, Maemura K, Nakasone H, Sakamoto K, Hirata Y, Nagai R: Takayasu myocarditis mediated by cytotoxic T lymphocytes. Intern Med 2005, 44: 256-260. 10.2169/internalmedicine.44.256
Eck M, Greiner A, Kandolf R, Schmausser B, Marx A, Muller-Hermelink HK: Active fulminant myocarditis characterized by T-lymphocytes expressing the gamma-delta T-cell receptor: a new disease entity? Am J Surg Pathol 1997, 21: 1109-1112. 10.1097/00000478-199709000-00017
Huber S, Lodge P: Coxsackievirus B3 myocarditis in Balb/c mice: evidence for autoimmunity to myocyte antigens. Am J Path 1984, 116: 21-26.
Huber S, Weller A, Herzum M, Lodge P, Estrin M, Simpson K, Guthrie M: Immunopathogenic mechanisms in experimental picornavirus-induced autoimmunity. Immunopathol Res 1988, 7: 279-291. 10.1159/000157123
Huber S, Lodge P, Herzum M, Estrin M, Olszewski J: The role of T lymphocytes in the pathogenesis of coxsackievirus B3 myocarditis. In Cardiomyopathy Update 1: Pathogenesis of Myocarditis and Cardiomyopathy. Edited by: Kawai C, Abelmann W, Matsumori A. Tokyo: University of Tokyo Press; 1987:9-21.
Torgerson TR: Regulatory T cells in human autoimmune diseases. Springer Semin Immunopathol 2006, 28: 63-76. 10.1007/s00281-006-0041-4
Sakaguchi S, Yamaguchi T, Nomura T, Ono M: Regulatory T cells and immune tolerance. Cell 2008, 133: 775-787. 10.1016/j.cell.2008.05.009
Sakaguchi S: Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol 2005, 6: 345-352. 10.1038/ni1178
Malek TR, Bayer AL: Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 2004, 4: 665-674. 10.1038/nri1435
Ray A, Khare A, Krishnamoorthy N, Qi Z, Ray P: Regulatory T cells in many flavors control asthma. Mucosal Immunol 2010, 3: 216-229. 10.1038/mi.2010.4
Ozdemir C, Akdis M, Akdis CA: T regulatory cells and their counterparts: masters of immune regulation. Clin Exp Allergy 2009, 39: 626-639. 10.1111/j.1365-2222.2009.03242.x
Wang SLJ, Wang M, Zhang J, Wang Z: Treatment and prevention of experimental autoimmune myocarditis with CD28 superagonists. Cardiology 2010, 115: 107-113. 10.1159/000256660
Frisancho-Kiss S, Nyland JF, Davis SE, Barrett MA, Gatewood SJ, Njoku DB, Cihakova D, Silbergeld EK, Rose NR, Fairweather D: Cutting edge: T cell Ig mucin-3 reduces inflammatory heart disease by increasing CTLA-4 during innate immunity. J Immunol 2006, 176: 6411-6415.
Van Houten N, Bouchard P, Moraska A, Huber S: Selection of an attenuated coxsackievirus B3 variant using a monoclonal antibody reactive to myocyte antigen. J Virol 1991, 65: 1286-1290.
Nowak M, Kopp F, Roelofs-Haarhuis K, Wu X, Gleichmann E: Oral nickel tolerance: Fas ligand-expressing invariant NK T cells promote tolerance induction by eliciting apoptotic death of antigen-carrying, effete B cells. J Immunol 2006, 176: 4581-4589.
Roelofs-Haarhuis K, Wu X, Gleichmann E: Oral tolerance to nickel requires CD4+ invariant NKT cells for the infectious spread of tolerance and the induction of specific regulatory T cells. J Immunol 2004, 173: 1043-1050.
Roelofs-Haarhuis K, Wu X, Nowak M, Fang M, Artik S, Gleichmann E: Infectious nickel tolerance: a reciprocal interplay of tolerogenic APCs and T suppressor cells that is driven by immunization. J Immunol 2003, 171: 2863-2872.
Sonoda KH, Exley M, Snapper S, Balk SP, Stein-Streilein J: CD1-reactive natural killer T cells are required for development of systemic tolerance through an immune-privileged site. J Exp Med 1999, 190: 1215-1226. 10.1084/jem.190.9.1215
La Cava A, Van Kaer L, Fu Dong S: CD4+CD25+ Tregs and NKT cells: regulators regulating regulators. Trends Immunol 2006, 27: 322-327. 10.1016/j.it.2006.05.003
Ly D, Mi QS, Hussain S, Delovitch TL: Protection from type 1 diabetes by invariant NK T cells requires the activity of CD4+CD25+ regulatory T cells. J Immunol 2006, 177: 3695-3704.
Cardell SL: The natural killer T lymphocyte: a player in the complex regulation of autoimmune diabetes in non-obese diabetic mice. Clin Exp Immunol 2006, 143: 194-202. 10.1111/j.1365-2249.2005.02942.x
Hong S, Wilson MT, Serizawa I, Wu L, Singh N, Naidenko OV, Miura T, Haba T, Scherer DC, Wei J, et al.: The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med 2001, 7: 1052-1056. 10.1038/nm0901-1052
Sonoda KH, Faunce DE, Taniguchi M, Exley M, Balk S, Stein-Streilein J: NK T cell-derived IL-10 is essential for the differentiation of antigen-specific T regulatory cells in systemic tolerance. J Immunol 2001, 166: 42-50.
Stein-Streilein J, Sonoda KH, Faunce D, Zhang-Hoover J: Regulation of adaptive immune responses by innate cells expressing NK markers and antigen-transporting macrophages. J Leukoc Biol 2000, 67: 488-494.
McGuirk P, Mills KH: Pathogen-specific regulatory T cells provoke a shift in the Th1/Th2 paradigm in immunity to infectious diseases. Trends Immunol 2002, 23: 450-455. 10.1016/S1471-4906(02)02288-3
Kumanogoh A, Wang X, Lee I, Watanabe C, Kamanaka M, Shi W, Yoshida K, Sato T, Habu S, Itoh M, et al.: Increased T cell autoreactivity in the absence of CD40-CD40 ligand interactions: a role of CD40 in regulatory T cell development. J Immunol 2001, 166: 353-360.
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA: B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000, 12: 431-440. 10.1016/S1074-7613(00)80195-8
Bach JF, Bendelac A, Brenner MB, Cantor H, De Libero G, Kronenberg M, Lanier LL, Raulet DH, Shlomchik MJ, von Herrath MG: The role of innate immunity in autoimmunity. J Exp Med 2004, 200: 1527-1531. 10.1084/jem.20042110
Chen G, Han G, Wang J, Wang R, Xu R, Shen B, Qian J, Li Y: Natural killer cells modulate overt autoimmunity to homeostasis in nonobese diabetic mice after anti-CD3 F(ab')2 antibody treatment through secreting transforming growth factor-beta. Am J Pathol 2009, 175: 1086-1094. 10.2353/ajpath.2009.080488
Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, Prinz I, Hemmer B, Kuchroo VK, Oukka M, Korn T: gammadelta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity 2010, 33: 351-363. 10.1016/j.immuni.2010.08.013
Hahn YS, Ji XY, Woo SI, Choi YK, Song MS, Shin KS, Jin N, O'Brien RL, Born WK: Vgamma1+ gammadelta T cells reduce IL-10-producing CD4+CD25+ T cells in the lung of ovalbumin-sensitized and challenged mice. Immunol Lett 2008, 121: 87-92. 10.1016/j.imlet.2008.09.001
Gong G, Shao L, Wang Y, Chen CY, Huang D, Yao S, Zhan X, Sicard H, Wang R, Chen ZW: Phosphoantigen-activated V gamma 2V delta 2 T cells antagonize IL-2-induced CD4+CD25+Foxp3+ T regulatory cells in mycobacterial infection. Blood 2009, 113: 837-845. 10.1182/blood-2008-06-162792
Bendelac A, Rivera MN, Park SH, Roark JH: Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu Rev Immunol 1997, 15: 535-562. 10.1146/annurev.immunol.15.1.535
Carding SR, Egan PJ: Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol 2002, 2: 336-345. 10.1038/nri797
Acknowledgements
The work was supported by grants HL80594 and HL86549 from the National Institutes of Health, and grants from the Youth Grants of the NSFC in China (30800481), youth special science and technology foundation in Heilongjiang Province (QC07C84).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
WL intellectually summarized the role of CD1d in virus induced myocarditis. She participated in the experiment (NKT related) and was involved in drafting the manuscript. SAH was involved in most of the basic experiments being summarized in this review. She designed most of the experiments we mentioned and revised this manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liu, W., Huber, S.A. Cross-talk between cd1d-restricted nkt cells and γδ cells in t regulatory cell response. Virol J 8, 32 (2011). https://doi.org/10.1186/1743-422X-8-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-8-32