
Abstract
Background
Saussurea involucrata (Kar. et Kir.) (S. involucrate), is a rare traditional Chinese medicinal herb. Rutin and hispidulin as well as their metabolites are flavonoids of the flavonol type that abound in S. involucrata, which has been reported to inhibit nonoxidative advanced glycation end products which was involved in physiological inflammation. This study aims to investigate the role of 3,4-dihydroxytoluene (DHT), a metabolite of rutin, in inflammatory inhibition and its involved mechanism.
Methods
This study utilized lipopolysaccharide (LPS) stimulated murine macrophage cell line RAW 264.7 as inflammatory model. The inhibitory effects of DHT were evaluated by the expression level of several inflammation markers such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) in RAW264.7 after LPS treatment. In addition, underlying mechanisms, the activation of mitogen-activated protein kinases (MAPKs) and NF-κB, were also investigated.
Results
Our results showed that DHT significantly suppressed the LPS-induced production of nitric oxide (NO), iNOS, and COX-2 in a dose-dependent manner without cytotoxicity. DHT also reduced the generation of proinflammatory cytokines majorly in tumor necrosis factor (TNF)-α and minor in interleukin (IL)-1β and IL-6. In addition, LPS-stimulated I-κBα phosphorylation and degradation followed by translocation of the nuclear factor κB (NF-kB)-p65 from the cytoplasm to the nucleus were attenuated after DHT treatment.
Conclusions
Combined, the results suggest that DHT might exert anti-inflammatory effects in vitro in LPS stimulated RAW 264.7 macrophages and is potential in adjuvant treatment in inflammation disease.
Similar content being viewed by others


Background
Saussurea involucrata (Kar. et Kir.) (S. involucrata), known as the snow lotus, grows in the rocky habitats of mountains that reach or exceed an elevation of 2600 m in the Tian Shan and A’er Tai areas of China. In recent years, the wild population of S. involucrata has been facing the threat of depletion due to their particularly slow growth and excessively harvested from the wild for pharmaceutical preparations. S. involucrata is now nearly extinct and has been listed as a second grade national protected wild plant in China[1, 2]. Based on the theories of traditional Chinese medicine, S. involucrata has the effect of warming the kidneys, activating yang, expelling wind, eliminating dampness, inducing menstruation, and promoting blood circulation. In folk medicine, it is used to treat rheumatoid arthritis, impotence, irregular menses, coughs and colds, stomachache, and altitude sickness, among other illnesses[3, 4]. Rutin (3,3’,4’,5,7-pentahydroxyflavone-3-rhamnoglucoside) is a flavonoid of the flavonol type abundantly present in S. involucrata. It has been reported that rutin has several pharmacological properties, including antioxidant, anticarcinogenic, cytoprotective, antiplatelet, antithrombotic, vasoprotective, and cardioprotective properties[5–10] as well as neuroprotective[11]. In addition, it also has been demonstrated to have anticonvulsant properties in pentylenetetrazol models in rats and mice[12]. In the mention of antioxidative issue, our previous report showed rutin can ameliorated ischemic reperfusion injuries in the brain by reducing MDA level in plasma and elevating caspase-3 and PARP[13].
High oral doses of rutin (3,3’,4’,5,7-pentahydroxyflavone-3-rhamnoglucoside) protected diabetic rats against the formation of protein adducts in the skin[14]. Although the precise mechanism of this protection is not known, little or no dietary rutin is absorbed because gut microflora in the large intestine metabolize rutin to a variety of compounds that include quercetin and phenol derivatives such as 3,4-dihydroxyphenylacetic acid (DHPAA), 3,4-dihydroxytoluene (DHT), -hydroxyphenylacetic acid (HPAA), and 4-hydroxy-3-methoxyphenylacetic acid (homovanillic acid, HVA)[15–20]. Pashikanti et. al. reported that the rutin metabolites can efficiently and powerfully inhibit fluorescent and nonfluorescent belong to advanced glycation end products (AGEs)[21]. Since AGEs were an important source of reactive dicarbonyl species (RCS) involved in several pathophysiological conditions such as inflammation[22, 23], we hypothesized that the metabolites can reduce inflammatory response. More importantly, rutin can reduce inflammatory cytokines such as IL-1 from the peritoneal macrophages (pMf).[17] Most of the rutin that is consumed is likely to be decomposed into low-molecular-weight phenolic acids by the colonic microflora[24, 25]. However, it had not been investigated what kind of metabolites of rutin can have anti-inflammation effect. In addition, it was evidenced that most of the protective effects of flavonoids against oxidative stress in PC12 cells are continued despite biodegradation of the parent flavonoids. Taken together, this background has led us to examine the suppression of the inflammation effects of dietary supplements using rutin’s derives, such as DHPAA, DHT, HPAA, and HVA on LPS-induced inflammation in macrophage cells.
In this study, we utilized Lipopolysaccharide (LPS)-stimulated murine macrophage cell line RAW 264.7 as inflammatory model to test the effects of rutin derives. LPS can trigger groups of different toll-like receptors (TLR) especially TLR4 expressed on the cell surface of immune cells[26]. The binding of LPS to TLR4 triggers down-stream signaling cascades, including mitogen-activated protein kinases (MAPKs)[27] and the nuclear transcription factor kappa-B (NF-B) pathways[28], which lead to the production of inflammatory mediators from macrophages, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), interleukin-6 (IL-6), and nitric oxide (NO)[29]. Our results indicated DHT, the phenol derivative of rutin, inhibited the NO, TNF-α in LPS-stimulated macrophages through Iκ B-NF-κ B signaling rather than MAPK, ERK1/2, p38 and JNK1/2 pathways. These results provided a model of DHT for anti-inflammation usage.
Methods
Preparation of fractions
The wild plant of S. involucrata used in this study was a gift from Biopure Biotechnology (Changhua, Taiwan). Twenty grams of dried aerial and flower parts S. involucrate was extracted with 100 mL of methanol three times under reflux for 2 h. The methanol extracts (SI-1) were combined, and the solvent was evaporated in vacuum to give a deep brown syrup. The syrup was resuspended in water and then partitioned successively with pentane, ethyl acetate (SI-2) and n-butanol (SI-3) to leave a water layer (SI-4). The solvents were evaporated respectively, and the residues were used throughout this study.
Reverse-phase high-performance liquid chromatography (HPLC) analysis of flavonoids in S. involucrata
The determination of flavonoids from S. involucrata was carried out by HPLC with a photodiary detector. The HPLC system consisted of a Shimadzu LC-20AT solvent delivery system, equipped with a SPD-M20A photodiode array detector, set at 270 nm. Samples were injected with SiL-20A autosample to separate on the TSK-Gel ODS-100S column. The column was maintained at an ambient temperature of 25°C. The flow rate of the system was 1.0 mL/min. The mobile phase consisted of solvent A (0.3% formic acid) and solvent B (acetonitrile). The elution profile for A was 0-10 min, with a linear gradient change of 0-5%; 10-40 min, with a linear gradient change to 55%; and maintained for another 10 min with a post run time to equilibrate the column and for the baseline to return to the normal and initial working conditions[2].
Chemicals and reagents
3,4-dihydroxyphenylacetic acid (DHPAA), 3,4-dihydroxytoluene (DHT), 3-hydroxyphenylacetic acid (HPAA) and 4-hydroxy-3-methoxyphenylacetic acid (homovanillic acid, HVA) and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO). LPS (from Escherichia coli 0111:B4), mouse antibodies against mouse phospho-ERK1/2, phospho-JNK1/2, phospho-p38, and actin were purchased from Sigma (St. Louis, MO). Rabbit antibodies against mouse ERK1/2, JNK1, p38, phospho-IκB-α, iNOS, COX-2, and HRP-second antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). IL-1β, IL-6, and TNF-α ELISA kits were purchased from R&D Systems (Minneapolis, MN).
Cell culture
RAW 264.7 macrophages were obtained from the American Type Culture Collection (Rockville, MD). The cells were propagated in RPMI-1640 medium (Gibco Laboratories, Grand Island, NY) supplemented with 10% heat-inactivated fetal calf serum (Biological Industries Ltd, Kibbutz Beit Haemek, Israel), and 2 mM L-glutamine (Life Technologies, Carlsbad, CA) at 37°C in a 5% CO2 incubator.
Cell viability assays
Cell viability assay was performed by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay based on the conversion of MTT into formazan crystals by living cells, which determines mitochondrial activity. Cell was cultured in 96-well plate with 5000/well confluence. Twenty-four hours after treatment, after removing supernatant 100 μl MTT reagent was added for 1 h incubation. For measurement, 100 μl DMSO was added in each well follow by ELISA reader detection at 570 nm. Data are representative of three independent experiments.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed according to the previous study[30]. Briefly, 2 × 106 in 2 mL medium were seeded in 60 mm dishes for treatment. Six hours after treatment, 50 μL of biotinylated antibody and 50 μL of supernatant were added to a stripwell plate precoated with antimouse IL-1β, IL-6, and TNF-α antibodies and incubated at room temperature for 2 h. After 3 washes with washing buffer, 100 μL of diluted HRP-conjugated streptavidin concentrate was added to each well and the plate incubated at room temperature for 30 min. The washing process was repeated, then 100 μL of a premixed tetramethylbenzidine substrate solution was added to each well and the reaction developed at room temperature in the dark for 30 min. Following addition of 100 μL of stop solution to each well, the absorbance was read in a microplate reader at 450 nm. Data are representative of three independent experiments.
NO measurement
1×105 cells was seeded into 24-well plate (Nunc, USA) at the day before treatment. Twenty-four hours after treatment, the supernatant was collected and NO measurement was performed by Griess Reagent. Briefly, the supernatant (100 μl) was reacted with 1% sulfanilamide solution (50 μl) for 5 min in the dark. After incubation, 50 μl 0.1% NED solution was added and absorbance was measured at 550 nm. The concentration of NO was calculated by utilizing NaNO2 standard curve. Data are representative of three independent experiments.
Western blot assay
After treatment, the cells were collected and lysed at 4°C in lysis buffer (25 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2.5 mM EDTA, 2.5 mM EGTA, 20 mM NaF, 1 mM Na3VO4, 20 mM sodium β-glycerophosphate, 10 mM sodium pyrophosphate, 0.5% Triton X-100) containing protease inhibitor cocktail (Sigma, St. Louis, MO), then the whole cell lysate was separated by SDS-PAGE and electrotransferred to a PVDF membrane. The membranes were incubated for 1 h at room temperature in blocking solution (5% nonfat milk in phosphate buffered saline with 0.1% Tween 20), then incubated for 2 h at room temperature with specific primary antibody in blocking solution. After 3 washes in PBS with 0.1% Tween 20, the membrane was incubated for 1 h at room temperature with HRPconjugated secondary antibody in blocking buffer and developed using an enhanced chemiluminescence Western blot detection system.
Statistical analysis
All values are given as means ± S. E. Data analysis involved one-way ANOVA with subsequent Scheffé test. * and ** indicate a significant difference at the level of p < 0.05 and P < 0.01, respectively, compared to LPS alone.
Results and discussion
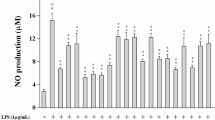
To test whether rutin and its’ metabolites can efficiently inhibit inflammation, we measured nitric oxide (NO) production, an inflammation marker, in LPS-stimulated RAW 264.7 cells in the present of these compounds. The result indicated 10 μM DHT had the most significant inhibitory effect on NO production when compared with other metabolites (Figure 1A). Therefore, we further utilized DHT to address its biological effects and mechanisms on inflammation. In the mention of cytotoxicity, the cell viability was over 80% after 10 μM rutin or metabolites treatment suggested that these compounds had low cytotoxicity. (Figure 1B). To evaluate the effect of DHT on NO production precisely, we performed NO measurement for RAW264.7 cell with different dose. The result indicated that DHT can significantly reduce NO generation after LPS treatment in a dose-dependent manner from 1.25 to 10 μM (Figure 2A). In addition to NO, we also test several inflammatory cytokines production after LPS stimulation including TNF-α, IL-6 and IL-1β. DHT at 5 and 10 mM can significantly reduce the mRNA and protein expression levels of TNF-# (Figure 2B). No significant effect was observed on IL-6 and IL-1b mRNA and protein expression (Figure 2C and D). Taken together, these results indicated that DHT can eliminate LPS-induced NO and inflammatory cytokines production majorly through TNF-α signaling without affecting cell viability.

Effects of rutin metabolites on NO production in LPS-stimulated RAW 264.7 and DHT cytotoxicity test. (A) After LPS stimulation, RAW264.7 macrophage with rutin or its metabolites including DHT, DHPAA, HPAA and HVA treatment was assessed to NO production measurement. (B) Cell cytotoxicity assay for DHT. RAW 264.7 was treated with DHT for 24 hours, followed by incubating with MTT reagent to measure OD 570 nm by spectrophotometry. The data was expressed as the means ± SE from three separated experiments.

Effect of DHT on inhibition nitric oxide and pro-inflammatory cytokines production in LPS-stimulated RAW264.7 macrophage. RAW264.7 macrophage were pre-treated with DHT for 30 min, followed by stimulating with LPS (100 ng/ml) for 6 and 24 hours. (A) After incubation for 24 h, nitric oxide concentration in culture medium was assayed by the Gress reaction. After incubation for 6 h, the (B) TNF-α and (C) IL-6 mRNA expression and secreted concentration in culture medium was assayed by RT-PCR and ELISA. (D) The levels of IL-1β mRNAs and protein were determined by RT-PCR and ELISsA. The data was expressed as the means ± SE from three separated experiments. *P < 0.05, **P < 0.01, significantly with the LPS-treated group only.
Since NO production can be catalyzed by inducible NO synthase (iNOS), we further investigated the effect of DHT on iNOS expression. The result showed that DHT can significantly reduce iNOS expression in a dose dependent manner. Similar results were obtained for cyclooxygenase-2 (COX-2) (Figure 3). These results indicated that the DHT was the principal metabolite of rutin with potential anti-inflammatory activity. MAPK signaling was one of the most important LPS-activated pathways, therefore, we tested whether DHT can modulate LPS-stimulated downstream signaling of MAPK. The results indicated that DHT had no effect on LPS mediated JNK1/2, ERK1/2 as well as p38 signaling (Figure 4A-B).

Effects of DHT on inhibition of COX-2 and iNOS expression in LPS-stimulated RAW264.7 macrophage. RAW264.7 macrophage were pre-treated with DHT for 30 min, followed by stimulating with LPS (100 ng/ml) for 24 hours. The protein levels of COX-2, iNOS and GAPDH were determined by Western blot analysis. (upper panel) RI (Relative intensity) was measured by the densitometry scanning (lower panel). The data was expressed as the means ± SE from three separated experiments. *P < 0.05, **P < 0.01, significantly with the LPS-treated group only.

Effects of DHT on activation of MAPK pathway in LPS-stimulated RAW264.7 macrophage. RAW264.7 macrophage were pre-treated with DHT for 30 min, followed by stimulating with LPS (100 ng/ml) for 30 min. (A) The phosphorylation and total protein levels of MAPK (ERK1/2, JNK1/2 and p38) were determined by Western blot analysis. (B) Quantification of the level of phosphorylation measured by the densitometry scanning. The data was expressed as RI (relative intensity) and the means ± SE from three separated experiments. *P < 0.05, **P < 0.01, significantly with the LPS-treated group only.
The NF-κB, one of the critical transcription factors, regulates the inflammatory-related gene expression in the LPS-activated macrophages. In the resting macrophages, NF-κB is sequestered in the cytoplasm as an inactive precursor complex by its inhibitory protein, IκB. After stimulation, IκB is phosphorylated by IκB kinase, ubiquitinated, and rapidly degraded through proteasomes followed by NF-κB releasing. Released p65 subunit of NF-κB is translocated from cytoplasm to nucleus and activates numerous corresponding genes. We found that DHT can significantly reduce the phosphorylation of IκB-a in RAW 264.7 cells after LPS treatment (Figure 5A). This leaded to accumulation of NF-κB (p65) in the cytoplasm and nuclear NF-κB (p65) reduction (Figure 5B). These results indicated that DHT at least can attenuate NF-κB signaling activation in macrophages after LPS treatment. Taken together, our results showed that DHT can efficiently inhibit IκB-NFκB mediated inflammatory response (Figure 6).

Effect of DHT on activation of NF-kB pathway in LPS-stimulated RAW264.7 macrophage. RAW264.7 macrophage were pre-treated with DHT for 30 min, followed by stimulating with LPS (100 ng/ml) for 30 min. (A) The phosphorylation and total protein levels of IκB and α-tubulin were determined by Western blot analysis. (B) The p65 subunit of NF-kB in cytoplasm and p65 in nucleus were determined by western blot analysis. RI (Relative intensity) was measured by the densitometry scanning. The data was expressed as the means ± SE from three separated experiments. *P < 0.05, **P < 0.01, significantly with the LPS-treated group only.

Illustration of signaling for RAW264.7 macrophage anti-inflammatory effect by DHT.
Inflammation not only participates in host defense but also controls the pathogenesis of numerous diseases including type 2 diabetes mellitus[31–34], atherosclerosis[35, 36], obesity[31, 36], silicosis[37], Alzheimer's disease[38], gout[33, 39] and kidney disease[40, 41]. An elevated level of circulating inflammatory cytokines, in particular, and a combined elevation of IL-1 and IL-6, constitute risk factors for the pathogenesis of type II diabetes mellitus[42]. Also, TNF-α was found in some metabolic diseases[43, 44]. Some genetic engineering animal model further confirmed inflammatory progress had impact in vascular disease[45]. Therefore, reduction of inflammation may provide an alternative disease management in complementary medicine field.
In this study, we found that rutin and DHT, the phenol derivative of rutin, can significantly reduce NO production in inflammatory response of LPS-stimulated macrophages. Although NO played a critical role in the host defense on various pathogens, the overproduction of NO can be harmful and result in septic shock, rheumatoid arthritis, and autoimmune diseases[46]. Therefore, adjuvant therapeutic agents that inhibit iNOS might be useful in relieving these inflammatory conditions. In this study, DHT can potentially inhibit iNOS as well as COX-2 expression through its upstream signaling in LPS-stimulated macrophages. The dual intrinsic enzyme activities of COX-2 catalyze 2 sequential reactions in the metabolism of arachidonic acid (AA). In addition, the COX-2 enzyme possesses a heme-containing active site that provides peroxidase activity, which requires 2 electrons (2e−) to become active. The peroxidase reaction converts PGG2 to PGH2 by removing oxygen(s), (Ox), which might be a source of oxygen radicals. Therefore, as more AA is metabolized to PG by COX-2, more electron donors are depleted and more oxygen radicals are generated. The effect of DHT on COX-2 expression also supported its potential role and application in physiological inflammatory unbalance by health supplements.
The reduction of iNOS and COX-2 may be due to transcription suppression. Among transcription factors, NF-κ B is a significant inflammation-related one for downstream gene activation including cytokines, chemokines, adhesion molecules, and acute phase proteins[47]. In addition, NF-κ B is a redox-sensitive transcription factor that regulates a multitude of inflammatory genes, including cytokines, chemokines, adhesion molecules, and acute phase proteins. Under basal conditions, NF-κ B is inactive by associated with inhibitory proteins in the cytoplasm to prevent DNA binding for transcription activity. In our study, after the treatment with DHT, the nucleic NF-κ B p65 was reduced while cytoplasmic one was accumulated (Figure 5). Although the reduction of iNOS and COX-2 may be the consequence of NF-κ B inactivation, other possible mechanisms involved in DHT effects can’t be excluded due to wide contribution of NF-κ B. Therefore, DHT may affect the upstream molecular of Iκ B-NF-κ B signaling, since the phosphorylation of Iκ B was attenuated by DHT. To clarify the underlying molecular networks, more experiments should be further performed. On the other hand, in this study, we found DHT had no influence on MAPK pathway (Figure 4) suggested DHT reduced TNF-α expression majorly through PI3K/AKT followed by NF-κ B activation.
Conclusion
We demonstrated the anti-inflammatory activity of DHT, the phenol derivative of rutin, via affecting Iκ B-NF-κ B signaling in macrophages and dissected its possible mechanism. The anti-inflammatory activity might be a basis for therapeutic supplements innovation in complementary and alternative medicine.
References
Fu LK: China plant Red data book: rare and endangered plants: vol 001. Science Pr. 1992
Way TD, Lee JC, Kuo DH, Fan LL, Huang CH, Lin HY, Shieh PC, Kuo PT, Liao CF, Liu H: Inhibition of epidermal growth factor receptor signaling by Saussurea involucrata, a rare traditional Chinese medicinal herb, in human hormone-resistant prostate cancer PC-3 cells. J Agric Food Chem. 2010, 58 (6): 3356-3365. 10.1021/jf903793p.
Liu JH, Ho SC, Lai TH, Liu TH, Chi PY, Wu RY: Protective effects of Chinese herbs on D-galactose-induced oxidative damage. Methods Find Exp Clin Pharmacol. 2003, 25 (6): 447-452. 10.1358/mf.2003.25.6.769650.
Chen YL, Lin SZ, Chang JY, Cheng YL, Tsai NM, Chen SP, Chang WL, Harn HJ: In vitro and in vivo studies of a novel potential anticancer agent of isochaihulactone on human lung cancer A549 cells. Biochem Pharmacol. 2006, 72 (3): 308-319. 10.1016/j.bcp.2006.04.031.
La Casa C, Villegas I, Alarcon de la Lastra C, Motilva V, Martin Calero MJ: Evidence for protective and antioxidant properties of rutin, a natural flavone, against ethanol induced gastric lesions. J Ethnopharmacol. 2000, 71 (1–2): 45-53.
Janbaz KH, Saeed SA, Gilani AH: Protective effect of rutin on paracetamol- and CCl4-induced hepatotoxicity in rodents. Fitoterapia. 2002, 73 (7–8): 557-563.
Schwedhelm E, Maas R, Troost R, Boger RH: Clinical pharmacokinetics of antioxidants and their impact on systemic oxidative stress. Clin Pharmacokinet. 2003, 42 (5): 437-459. 10.2165/00003088-200342050-00003.
Sheu JR, Hsiao G, Chou PH, Shen MY, Chou DS: Mechanisms involved in the antiplatelet activity of rutin, a glycoside of the flavonol quercetin, in human platelets. J Agric Food Chem. 2004, 52 (14): 4414-4418. 10.1021/jf040059f.
Mellou F, Loutrari H, Stamatis H, Roussos C, Kolisis FN: Enzymatic esterification of flavonoids with unsaturated fatty acids: effect of the novel esters on vascular endothelial growth factor release from K562 cells. Process Biochem. 2006, 41 (9): 2029-2034. 10.1016/j.procbio.2006.05.002.
Trumbeckaite S, Bernatoniene J, Majiene D, Jakštas V, Savickas A, Toleikis A: The effect of flavonoids on rat heart mitochondrial function. Biomed Pharmacother. 2006, 60 (5): 245-248. 10.1016/j.biopha.2006.04.003.
Katsube T, Imawaka N, Kawano Y, Yamazaki Y, Shiwaku K, Yamane Y: Antioxidant flavonol glycosides in mulberry (Morus alba L.) leaves isolated based on LDL antioxidant activity. Food Chem. 2006, 97 (1): 25-31. 10.1016/j.foodchem.2005.03.019.
Nassiri-Asl M, Shariati-Rad S, Zamansoltani F: Anticonvulsive effects of intracerebroventricular administration of rutin in rats. Progr Neuro Psychopharmacol Biol Psychiatr. 2008, 32 (4): 989-993. 10.1016/j.pnpbp.2008.01.011.
Gupta R, Singh M, Sharma A: Neuroprotective effect of antioxidants on ischaemia and reperfusion-induced cerebral injury. Pharmacol Res. 2003, 48 (2): 209-215. 10.1016/S1043-6618(03)00102-6.
Kuo SM: Antiproliferative potency of structurally distinct dietary flavonoids on human colon cancer cells. Cancer Lett. 1996, 110 (1–2): 41-48.
Middleton E: Effect of plant flavonoids on immune and inflammatory cell function. Adv Exp Med Biol. 1998, 439: 175-182. 10.1007/978-1-4615-5335-9_13.
Deschner EE, Ruperto J, Wong G, Newmark HL: Quercetin and rutin as inhibitors of azoxymethanol-induced colonic neoplasia. Carcinogenesis. 1991, 12 (7): 1193-1196. 10.1093/carcin/12.7.1193.
Kwon KH, Murakami A, Ohigashi H: Suppressive effects of natural and synthetic agents on dextran sulfate sodium-induced interleukin-1beta release from murine peritoneal macrophages. Biosci Biotechnol Biochem. 2004, 68 (2): 436-439. 10.1271/bbb.68.436.
Braune A, Gutschow M, Engst W, Blaut M: Degradation of quercetin and luteolin by Eubacterium ramulus. Appl Environ Microbiol. 2001, 67 (12): 5558-5567. 10.1128/AEM.67.12.5558-5567.2001.
Griffiths LA, Barrow A: Metabolism of flavonoid compounds in germ-free rats. Biochem J. 1972, 130 (4): 1161-1162.
Schneider H, Simmering R, Hartmann L, Pforte H, Blaut M: Degradation of quercetin-3-glucoside in gnotobiotic rats associated with human intestinal bacteria. J Appl Microbiol. 2000, 89 (6): 1027-1037. 10.1046/j.1365-2672.2000.01209.x.
Pashikanti S, de Alba DR, Boissonneault GA, Cervantes-Laurean D: Rutin metabolites: novel inhibitors of nonoxidative advanced glycation end products. Free Radic Biol Med. 2010, 48 (5): 656-663. 10.1016/j.freeradbiomed.2009.11.019.
Anderson MM, Hazen SL, Hsu FF, Heinecke JW: Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. A mechanism for the generation of highly reactive alpha-hydroxy and alpha,beta-unsaturated aldehydes by phagocytes at sites of inflammation. J Clin Invest. 1997, 99 (3): 424-432. 10.1172/JCI119176.
Lee W, Ku S-K, Bae J-S: Barrier protective effects of rutin in LPS-induced inflammation in vitro and in vivo. Food Chem Toxicol. 2012, 50 (9): 3048-3055. 10.1016/j.fct.2012.06.013.
Olthof MR, Hollman PC, Buijsman MN, van Amelsvoort JM, Katan MB: Chlorogenic acid, quercetin-3-rutinoside and black tea phenols are extensively metabolized in humans. J Nutr. 2003, 133 (6): 1806-1814.
Winter J, Moore LH, Dowell VR, Bokkenheuser VD: C-ring cleavage of flavonoids by human intestinal bacteria. Appl Environ Microbiol. 1989, 55 (5): 1203-1208.
Kaisho T, Akira S: Critical roles of Toll-like receptors in host defense. Crit Rev Immunol. 2000, 20 (5): 393-405.
Sweet MJ, Hume DA: Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996, 60 (1): 8-26.
Doyle SL, O’Neill LAJ: Toll-like receptors: from the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol. 2006, 72 (9): 1102-1113. 10.1016/j.bcp.2006.07.010.
Takeda K, Kaisho T, Akira S: Toll-like receptors. Annu Rev Immunol. 2003, 21: 335-376. 10.1146/annurev.immunol.21.120601.141126.
Liao P-C, Chien S-C, Ho C-L, Wang EIC, Lee S-C, Kuo Y-H, Jeyashoke N, Chen J, Dong W-C, Chao LK: Osthole regulates inflammatory mediator expression through modulating NF-κB, mitogen-activated protein kinases, protein kinase C, and reactive oxygen species. J Agric Food Chem. 2010, 58 (19): 10445-10451. 10.1021/jf102812t.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD: The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011, 17 (2): 179-188. 10.1038/nm.2279.
Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z: Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010, 11 (10): 897-904. 10.1038/ni.1935.
Schroder K, Zhou R, Tschopp J: The NLRP3 inflammasome: a sensor for metabolic danger?. Science. 2010, 327 (5963): 296-300. 10.1126/science.1184003.
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP: Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011, 12 (5): 408-415. 10.1038/ni.2022.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M: NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010, 464 (7293): 1357-1361. 10.1038/nature08938.
De Nardo D, Latz E: NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011, 32 (8): 373-379. 10.1016/j.it.2011.05.004.
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E: Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008, 9 (8): 847-856. 10.1038/ni.1631.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT: The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008, 9 (8): 857-865. 10.1038/ni.1636.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006, 440 (7081): 237-241. 10.1038/nature04516.
Anders H-J, Muruve DA: The inflammasomes in kidney disease. J Am Soc Nephrol. 2011, 22 (6): 1007-1018. 10.1681/ASN.2010080798.
Tsai P-Y, Ka S-M, Chang J-M, Chen H-C, Shui H-A, Li C-Y, Hua K-F, Chang W-L, Huang J-J, Yang S-S: Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic Biol Med. 2011, 51 (3): 744-754. 10.1016/j.freeradbiomed.2011.05.016.
Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, Boeing H, Pfeiffer AFH: Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based european prospective investigation into cancer and nutrition (EPIC)-potsdam study. Diabetes. 2003, 52 (3): 812-817. 10.2337/diabetes.52.3.812.
Yehuda H, Szuchman-Sapir A, Khatib S, Musa R, Tamir S: Human atherosclerotic plaque lipid extract promotes expression of proinflammatory factors in human monocytes and macrophage-like cells. Atherosclerosis. 2011, 218 (2): 339-343. 10.1016/j.atherosclerosis.2011.07.120.
Janabi M, Yamashita S, Hirano K-i, Sakai N, Hiraoka H, Matsumoto K, Zhang Z, Nozaki S, Matsuzawa Y: Oxidized LDL–induced NF-κB activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from CD36-deficient patients. Arterioscler Thromb Vasc Biol. 2000, 20 (8): 1953-1960. 10.1161/01.ATV.20.8.1953.
Kleemann R, Zadelaar S, Kooistra T: Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008, 79 (3): 360-376. 10.1093/cvr/cvn120.
Lee RP, Subeq YM, Lee CJ, Hsu BG, Peng TC: Freshwater clam extract decreased hemorrhagic shock-induced liver injury by attenuating TNF-alpha production. Biol Res Nurs. 2012, 14 (3): 286-293. 10.1177/1099800411408881.
Baeuerle PA: IκB–NF-κB structures: at the interface of inflammation control. Cell. 1998, 95 (6): 729-731. 10.1016/S0092-8674(00)81694-3.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6882/14/21/prepub
Acknowledgements
This work was supported by the National Science Council, Taiwan: contract grant numbers: 98-2320-B-197-002-MY3 (Y. L. S. Chen).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YLC made contributions to conception, design and gave final approval of the manuscript to be published. YPC performed experiments and collected the data for analysis. KFH and KYS provided the interpretation of data and were involved in drafting the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Su, KY., Yu, C.Y., Chen, YP. et al. 3,4-Dihydroxytoluene, a metabolite of rutin, inhibits inflammatory responses in lipopolysaccharide-activated macrophages by reducing the activation of NF-κB signaling. BMC Complement Altern Med 14, 21 (2014). https://doi.org/10.1186/1472-6882-14-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6882-14-21