
Abstract
Background
In the anaerobic pathway of cobalamin (vitamin B12) synthesis, the CbiT enzyme plays two roles, as a cobalt-precorrin-7 C15-methyltransferase and a C12-decarboxylase, to produce the intermediate, cobalt-precorrin 8.
Results
The primary structure of the hypothetical protein MJ0391, from Methanocaldococcus jannaschii, suggested that MJ0391 is a putative CbiT. Here, we report the crystal structure of MJ0391, solved by the MAD procedure and refined to final R-factor and R-free values of 19.8 & 27.3%, respectively, at 2.3 Å resolution. The asymmetric unit contains two NCS molecules, and the intact tetramer generated by crystallographic symmetry may be functionally important. The overall tertiary structure and the tetrameric arrangements are highly homologous to those found in MT0146/CbiT from Methanobacterium thermoautotrophicum.
Conclusions
The conservation of functional residues in the binding site for the co-factor, AdoMet, and in the putative precorrin-7 binding pocket suggested that MJ0391 may also possess CbiT activity. The putative function of MJ0391 is discussed, based on structural homology.
Similar content being viewed by others


Background
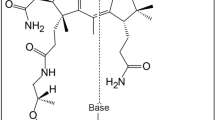
The de novo synthesis of vitamin B12 (cobalamin) occurs via a complex biochemical pathway involving more than 30 enzyme-mediated steps [1, 2]. Therefore, many organisms do not use cobalamin as a coenzyme, and neither synthesize nor require the vitamin in their metabolism. Interestingly, only certain eubacteria and archaea synthesize cobalamin, and the pathway apparently never made the transition to eukaryotes [3]. Although they are genetically distinct, two related pathways are involved in cobalamin biosynthesis in prokaryotes: the aerobic and anaerobic pathways [4]. The aerobic pathway requires molecular oxygen and is characterized by the comparatively late chelation of cobalt into the corrin ring [2, 5–8]. In contrast, the anaerobic pathway does not require molecular oxygen, and the cobalt is inserted at an early synthetic stage [4, 9, 10]. For example, the pathway in Pseudomonas denitrificans, an aerobe, incorporates molecular oxygen into the precorrin-3 macrocycle as a prerequisite to ring contraction, and hence is classified as an aerobic pathway. The pathway in Salmonella typhimurium synthesizes cobalamin only in the absence of molecular oxygen. This pathway utilizes cobalt ion chelated into precorrin-2 (or Factor II) to set the stage for ring contraction, and hence is classified as anaerobic. The biosynthesis of precorrin-2 is common to both the aerobic and anaerobic pathways. At this stage, the two pathways divide, rejoining only after the synthesis of Cob(II)yrinate a, c diamide (Figure 1) [1].

The aerobic and anaerobic pathways of cobalamin synthesis. CobL in the aerobic pathway is colored red. CbiE and CbiT, which are equivalent to CobL, are colored blue and red with a boxed label, respectively, in the anaerobic pathway. The chemical structures of cobalt-precorrin-6B, 7 and 8, which are the substrates and products of CbiE and CbiT, are shown at the bottom of the figure.
The intermediate steps of the aerobic biosynthetic pathway, from uroporphyrinogen III (urogen III) to adenosylcobalamin, have been elucidated by the over-expression of the cob genes from P. denitrificans (for recent reviews, see [2, 11]), and the related assignments of the functions of the encoded enzymes, based on their catalytic activities. The detailed intermediate mechanisms of the anaerobic pathway for cobalamin biosynthesis are progressively emerging, through combinations of molecular genetic, biochemical and three-dimensional structural studies. The cbi genes involved in the biosynthesis of cobalamin by the anaerobic pathway have been over-expressed from not only S. typhimurium but also Bacillus megaterium [12], Propionibacterium freudenreichi (P. shermanii) [13] and Methanocaldococcus jannaschii [14].
The functions of CbiA, -B, -C, -D, -E, -F, -G, -H, -J, -K(X), -L, -P, and -T in the anaerobic pathway were assigned, mainly based on the sequence similarity to their counterparts in the corresponding, well characterized aerobic pathway of P. denitrificans (Figure 1). Experimental evidence for the functions of CbiB, -H, -K, and –L, and for those of CbiA, -D, -F, -G, and –T, among the 13 catalytic Cbi proteins, was also recently reported [15–17]. However, only the crystal structures of CbiC [18], CbiF [19], CbiK [20], CbiL [21] and CbiT [22] have been solved.
In the aerobic pathway, the enzyme CobL catalyzes the methylations at C-5 and C-15, and the decarboxylation of the C-12 acetate side chain of precorrin-6B. In contrast, in the anaerobic system, the functions of CobL are shared by two separate enzymes, CbiE and CbiT. Based on the sequence similarity, it has long been assumed that CbiE and CbiT are the methyltransferase and the decarboxylase, respectively: CbiE catalyzes the addition of the two methyl groups to cobalt-precorrin-6B, and CbiT then decarboxylates the C-12 acetate side chain to afford cobalt-precorrin 8. However, recent studies based on structural [22] and biochemical analyses revealed that CbiT possesses both the decarboxylase and methyltransferase activities; i.e., CbiT alone can catalyze both the methylation at C-15 and the decarboxylation of the C-12 acetate side chain, even in the absence of C-1 methylation [16].
The sequence analysis of the hypothetical protein MJ0391, from M. jannaschii (hereafter, referred to as MJ0391), suggested that it is closely related to MT0146/CbiT, with 33% sequence identity. Our crystal structure analysis of MJ0391, determined by the multiple anomalous dispersion (MAD) method, suggested that MJ0391 may possess the putative dual functional activities of decarboxylation and methyltransfer in the cobalamin biosynthesis pathway.
Results and discussion
Overall structure of MJ0391
The overall tertiary structure of the MJ0391 protein possesses the Rossmann-like “AdoMet-dependent methyltransferase” fold, comprising seven β-strands (Figure 2a). A long flexible stretch with a non-specific secondary structure conformation is observed at the N-terminal region, Met4 – Thr19. The methyltransferase fold is very similar to a canonical Rossmann fold, but it includes an antiparallel strand (β8) between the parallel strands, β5 and β6, at the C-terminal region. Furthermore, this fold is modified by the extension of the β6 strand, which forms a long irregular β hairpin with the β7 and β8 strands (Figure 2a). The canonical class I Rossmann-like methyltransferase fold consists of a central twisted seven-stranded β-sheet (β3-β2-β1-β4-β5-β8-β6), flanked by two bundles of helices on both sides. All of the strands are parallel except for the β8 strand, which is antiparallel to the other strands. Since the structure of MJ0391 is quite similar to that of MT0146/CbiT [22], the same nomenclature has been used for its description.

The structure of MJ0391. (a) Ribbon diagram (stereo view) of the tertiary structure of MJ0391 (colored in a rainbow ramp from blue at the N-terminus to red at the C-terminus). (b) The ribbon diagram of the MJ0391 dimer in the asymmetric unit (shown in an arbitrary orientation). Subunit A is colored in a rainbow ramp as in (a), and subunit B is colored green. In the dimer, β6 of subunit A and β6’ of subunit B form an intermolecular antiparallel β-sheet. The MES molecule is shown as a stick model. All figures were produced with the PyMOL program (Schrödinger, LLC.), unless otherwise mentioned. (c) The quaternary structure of MJ0391. Stereo view of the intact tetramer arrangement of MJ0391, produced by the crystallographic 2-fold axis. The tetramer is shown in an arbitrary orientation. Subunits A-D are colored red, green, orange and blue, respectively. A few structural elements at the tetramer interface are labeled, for clarity.
Oligomeric structure
The asymmetric unit contains two MJ0391 molecules (subunits A and B). The two subunits are stabilized by the formation of intermolecular antiparallel β-sheets between β6 and β6’ (Figure 2b). The crystal packing analysis revealed that the MJ0391 protein forms an intact tetramer (dimer of dimers) through the crystallographic 2-fold axis (Figure 2c). Each subunit of the tetramer contributes approximately 27 intermolecular electrostatic interactions to facilitate tetramerization (Table 1). In addition to these direct intermolecular interactions, several indirect hydrogen bond interactions are also formed through buried water molecules at the tetramer interface. The long, irregular β-hairpin, which is formed between the β6 and β7/β8 strands, plays a critical role in the tetramer formation (hereafter, referred to as the tetramerization hairpin (TH) region). The tetramerization occurs due to the contributions of the TH regions of each subunit, A-D. The TH region of subunit A forms intermolecular interactions with subunits B and C (Figure 2c). The β6 strand of subunit A forms an antiparallel sheet with β6’ of subunit B (Figure 2b), while the region connecting the β7 and β8 of subunit A strands forms intermolecular interactions with the corresponding region in subunit C (Figure 2c).
An intermolecular twisted β-sheet is formed between β6 and β6’ of subunits A and B, respectively. The residues Ala153, Asn155, Phe157, Ser159, and Ala161 within β6 of subunit A contribute intermolecular main-chain hydrogen bond interactions with the residues Ala161, Ser159, Phe157, Asn155, and Ala153 of subunit B, respectively (Table 1). Moreover, the residues His168 and Ala172 of subunit A, which are located in β7, form main-chain hydrogen bonds with Leu134 (α6) and Ala172 (β7), respectively, of subunit C.
The α1 helix also contacts the corresponding helix of its dimer counterpart. The subunit A residues Glu21 and Glu22 form potential salt links with Lys30 of subunit B. An additional salt link is also observed between Lys30 of subunit A and Glu22 of subunit B (Table 1). The α6 helix of subunit A potentially interacts with subunits B, C and D (Figure 2c). In subunit A, Glu135 electrostatically interacts with His168 and Asn142 of subunits C and D, respectively, while Asn142 of subunit A forms a salt-link with Glu135 of subunit D. Moreover, the Glu145 residue of subunit A also electrostatically interacts with Lys163 and His168 of subunit B. Besides these electrostatic interactions, about 17 residues of subunit A contribute substantial hydrophobic interactions toward tetramer formation (Table 2). The homologous protein MT0146/CbiT forms a tetramer in the crystal structure and in solution [22]. The buried surface area per monomer within the intact tetramer is about 1,907 Å2 (~21% buried surface area), as calculated by the program SURFACE in the CCP 4 suite. Since the overall tetramer arrangement in the MJ0391 structure is very similar to that found in MT0146/CbiT, we speculate that in solution, MJ0391 may also form a tetramer that is functionally important for its biological activity.
Comparison with homologous structures
A DALI [23] search revealed that although MJ0391 shares less sequence similarity with MT0146/CbiT from Methanobacterium thermoautotrophicum (33% identity; Z-score: 27.3), the precorrin-6Y C5, C15 – methyltransferase from Geobacter metallireducens GS-15 (Q39YF0; 32% identity; Z-score: 26.0), and the putative precorrin-6Y C5, C15 – methyltransferase from Corynebacterium diphtheriae (Q6NHA4; 17% identity; Z-score: 17.9) (Figure 3a), the overall tertiary structures and the quaternary structure arrangements are essentially conserved between them. The superimposition of the MJ0391 structure onto the structures of MT0146/CbiT (PDB ID: 1L3I), Q39YF0 (PDB ID: 3E05; 211-405aa), and Q6NHA4 (PDB ID: 3HM2; 236-410aa) yielded r.m.s.d. values of 1.6, 1.7, and 1.9 Å, respectively, for all Cα atoms (Figure 3b). Recently, the crystal structure of the C-terminal region of CobL (CobLc) from Rhodobacter capsulatus was reported [24]. The enzyme CobL, which uses precorrin-6B as a substrate, promotes the decarboxylation of the acetic acid side chain at C12, and the methylation at the C5 & C15 meso positions, to produce precorrin-8X. The superimposition of MJ0391 onto the CobLc structure (PDB ID: 3NJR) yielded Z-score and r.m.s.d. values of 25.6 and 1.8 Å, respectively, for 173 Cα atoms (Figure 3b).

Comparison with CbiT homologous proteins. (a) Sequence alignment of MJ0391 from Methanocaldococcus jannaschii, MT0146/CbiT from Methanobacterium thermoautotrophicum, precorrin-6Y C5, C15 – methyltransferase from Geobacter metallireducens GS-15 (Q39YF0), and the putative precorrin-6Y C5, C15 – methyltransferase from Corynebacterium diphtheriae (Q6NHA4). The secondary structural features from MJ0391 are indicated above the alignments. The colors reflect the similarity (red boxes and white characters for conserved residues; red characters for similarity in a group; blue frames for similarity across groups). The sequence was aligned and rendered by Clustal W [25] and ESPript [26], respectively. (b) Superposition of MJ0391 (yellow) on the precorrin-6Y C5, C15 – methyltransferase structures from G. metallireducens (PDB ID: 3E05, blue) and CobLc from R. capsulatus (PDB ID: 3NJR, cyan).
As expected, the DALI search with MJ0391 also identified proteins belonging to the methyltransferase family. For example, MJ0391 is structurally homologous to m1A58 tRNA methyltransferase, TrmI (PDB ID: 1O54, Z-score: 20.6, 2.9 Å r.m.s.d. for 174 Cα atoms, 12% identity), m5U1939 23S rRNA methyltransferase, RlmD (PDB ID: 2BH2, Z-score: 20.0, 2.6 Å r.m.s.d. for 169 Cα atoms, 16% identity), catechol O-methyltransferase, COMT (PDB ID: 1JR4, Z-score: 17.8, 2.3 Å r.m.s.d. for 161 Cα atoms, 17% identity), and rRNA dimethyladenosine transferase, Dim1 (PDB ID: 3FYC, Z-score: 18.7, 2.4 Å r.m.s.d. for 153 Cα atoms, 30% identity).
Comparison with MT0146/CbiT
Although the overall tertiary structures of the MJ0391 and MT0146/CbiT proteins are essentially similar, striking differences exist in the TH region, an important element for forming the tetrameric arrangements (Figure 4a). The dihedral angles at the equivalent positions of Lys163 (φ, ψ: -68, 140°) and Ala166 (φ, ψ: -69, 81°) in MJ0391 and MT0146/CbiT, respectively, trigger large deviations in their respective TH elements. Moreover, the β6 & β7 strands in MJ0391 are connected by a type II β-turn. The type II conformation is produced by incorporating Pro165 at the i+1 position. The β-turn is stabilized by an intra-hydrogen bond between the carbonyl-group of Ile164 (i position) and the amino-group of Gly167 (i+3 position). In contrast, in MT0146/CbiT, β6 and β7 are connected by a relatively long loop, comprising six amino acids. The abovementioned structural deviations cause the maximum r.m.s.d value of 6.2 Å between the Cα position of Pro165 in MJ0391 and that of Asp168 in MT0146. The α6 helix also participates in tetrameric interactions (Figure 2c); however, less structural deviation is observed between the corresponding regions of MJ0391 and MT0146/CbiT. In addition, deviations were observed in the non-specific secondary structure regions, such as the N-terminal region and the loops connecting the secondary structural elements (Figure 4a).

Comparison of MJ0391 with MT0146/CbiT. (a) Stereo view of the superposition of MJ0391 (light green) on MT0146/CbiT (red), for all Cα-atoms corresponding to one subunit of the MT0146/CbiT tetramer (PDB ID: 1L3I). (b) The AdoHcy binding site region (stereo view). The interacting residues and the AdoHcy molecule (shown as ‘SAH’ in the figure) of the MT0146/CbiT complex are colored grey & cyan, respectively. The equivalent predicted interacting residues in MJ0391 are yellow. The Tyr65 residue is modeled as Ala, since the electron density corresponding to its side chain was not visible. (c) Electrostatic surface potential of the MJ0391 tetramer. The surfaces are colored blue and red for positive and negative electrostatic surface potentials, denoting less than -15 and greater than +15 K bT, respectively, where K b is the Boltzmann constant and T is the temperature. The yellow and green arrows indicate the putative precorrin and AdoMet binding pockets, respectively. The surface potential was calculated with the software APBS[27] incorporated in the PyMOL program (Schrödinger, LLC.).
A comparison of the apo-form MJ0391 to the MT0146/CbiT complexed with AdoHcy (SAH molecule, S-Adenosyl-L-Homocysteine; PDB ID: 1L3I) [22] revealed that the co-factor binding site regions are similar in both structures (Figure 4b). Hence, we predict that the MJ0391 protein may also recognize the cofactor AdoMet in a similar manner as in MT0146/CbiT. The shallow binding pocket is produced by the head regions of β1, β2 and β4, which line the middle of the pocket. The pocket is surrounded by the α2 and α3 helices on one side, and the α4 and α5 helices on the other side (Figure 4b). The residues within the loops connecting β4 and α5, β2 and α3, β3 and α4, β1 and α2 and in the N-terminal flexible region may potentially form interactions with the AdoHcy molecule. The AdoHcy interacting residues are nearly conserved between these proteins, except for the residues Arg91, Tyr65, Lys110 and Asn111, which are replaced by Asp90, Arg63, Gly111 and Glu112, respectively, in MT0146/CbiT. These loops are quite flexible, as observed in Figure 4a, and thus we speculate that they may undergo conformational changes upon binding to the AdoHcy/AdoMet molecule.
Near the AdoMet binding site, a deep groove is formed by segments from the Rossmann-like methyltransferase core and the TH region (of the same subunit and of the adjacent subunit) (Figure 2c). Keller et al. [22] suggested that this deep groove is a putative binding pocket for the precorrin molecule. Surface potential analyses revealed that the MJ0391 protein is highly basic (theoretical pI: 9.01) (Figure 4c), whereas its homologous protein, MT0146/CbiT, is highly acidic (theoretical pI: 4.88). Although the electrostatic charge distributions are quite different between these two proteins, the charge distributions within the putative precorrin binding pocket are highly conserved between them (Figure 4c). The residues Pro17, Arg24, Lys162, Lys173 and Asn174 are located near the binding pocket in MJ0391. The equivalent corresponding residues in MT0146/CbiT are Pro14, Arg22, Arg165, Arg176 and Asn177, respectively (Figure 3). Recent studies of S. typhimurium CbiT revealed that CbiT alone can catalyze both the methylation at C15 and decarboxylation of the C12 acetate side chain, even in the absence of C1 methylation [16]. Since MJ0391 is structurally highly homologous to the known CbiT proteins, and the functional residues are conserved between them, we speculate that MJ0391 may also possess the dual roles of methylation and decarboxylation to form the intermediate, cobalt-precorrin 8, in the vitamin B12 biosynthesis pathway. However, further biochemical and crystallographic studies are required to understand the enzymatic mechanism of MJ0391 in cobalamin synthesis.
Conclusions
The structure of the hypothetical protein MJ0391, from M. jannaschii, was determined by the MAD procedure, which revealed the presence of a Rossmann-like “AdoMet-dependent methyltransferase” fold. The tertiary and quaternary structures of MJ0391 are essentially similar to those of the CbiT or CobLc proteins from M. thermoautotrophicum, G. metallireducens, and R. capsulatus. Moreover, the functional residues in the putative precorrin binding pocket are highly conserved. Hence, based on structural comparison studies, we speculate that the MJ0391 protein may possess the putative dual C15-methyltransferase and C12-decarboxylase activities in the cobalamin biosynthesis pathway.
Methods
Protein expression and purification
The gene encoding the MJ0391 protein (gi:15668567) was amplified via PCR using Methanocaldococcus jannaschii DSM 2661 genomic DNA, and was cloned into the pET-21a expression vector (Merck Novagen, Darmstadt, Germany). The expression vector was introduced into the E. coli BL21-CodonPlus (DE3)-RIL-X strain (Stratagene, La Jolla, California, USA). The recombinant strain was cultured in 3 L minimal medium, containing 25 μg/mL selenomethionine, 30 μg/mL chloramphenicol and 50 μg/mL ampicillin. The harvested cells (10.7 g) were lysed by sonication on ice, in 20 mL of 20 mM Tris–HCl buffer, pH 8.0, containing 500 mM NaCl, 5 mM β-mercaptoethanol and 1 mM phenylmethylsulfonyl fluoride. The cell lysate was heat-treated at 363 K for 11.5 min and centrifuged at 14,000 g for 30 min at 277 K. The supernatant was desalted by fractionation on a HiPrep 26/10 column (GE Healthcare Biosciences) with 20 mM Tris–HCl buffer, pH 8.0. The fractions of interest were then applied to a Super Q Toyopearl 650M column (Tosoh, Tokyo) equilibrated with 20 mM Tris–HCl buffer, pH 8.0, and eluted with a linear gradient of NaCl. The target fractions, which eluted in 0.1 M NaCl, were applied to a Resource Q column (GE Healthcare Biosciences) equilibrated with 20 mM Tris–HCl (pH 8.0). The flow-through fraction was collected and applied to a Hydroxyapatite CHT-I column (Bio-Rad Laboratories), equilibrated with 10 mM potassium phosphate buffer (pH 7.0), and eluted with a linear gradient of potassium phosphate. The target sample, which eluted in 207.2 mM potassium phosphate, was collected and applied to a HiLoad 16/60 Superdex 200pg column (GE Healthcare Biosciences), equilibrated with 20 mM Tris–HCl buffer, pH 8.0, containing 200 mM NaCl. The protein sample was analyzed by SDS-PAGE and was confirmed by N-terminal amino-acid sequencing. After concentration to 27.9 mg/mL by ultrafiltration, the protein yield was 11.7 mg from 10.7 g of cells.
Crystallization and data collection
Crystallization screening was performed by the sitting-drop vapor-diffusion method at 293 K. Each drop consisted of 1.0 μL of the Se-Met protein (28 mg/mL) solution and 1.0 μL of the reservoir solution, and was equilibrated against 100 μL of reservoir solution. In the preliminary screening, small crystals were observed in the crystallization conditions of 40%(v/v) MPD, 0.1 M Na HEPES, pH 7.0. After optimization, crystals suitable for X-ray data collection appeared in 50%(v/v) PEG-400, 0.1 M MES buffer, pH 6.0, within 2 months. The crystals were flash-cooled in a nitrogen-gas stream at 100 K, using 25%(v/v) glycerol as a cryoprotectant.
Anomalous diffraction data were collected at the SPring-8 (Harima, Japan) BL26B1 beamline, at three wavelengths, 0.97891 (peak), 0.97928 (edge) and 0.90000 Å (remote), for a Se-MAD experiment. The data sets were collected at 100 K, using a Rigaku Jupiter 210 CCD detector. The crystal-to-detector distance was set to 200 mm. The diffraction data were integrated, reduced, and scaled using the HKL2000 software suite [28]. The crystallographic data and refinement statistics are shown in Table 3. The asymmetric unit consists of two NCS molecules, with a molecular mass of 21 kDa and 183 amino acid residues per monomer, and with a solvent content of about 42%.
Structure determination and refinement
The structure determination was performed by the multiple anomalous dispersion (MAD) method, using SOLVE/RESOLVE [29]. The MAD procedure yielded six distinct peaks, corresponding to six of the eight selenium atoms in the asymmetric unit, with an overall experimental mean figure of merit of 0.62 for all data up to 2.5 Å resolution. The initial experimental phases were further improved by solvent flattening with 2-fold non-crystallographic symmetry (NCS) averaging, using the program RESOLVE [30], which increased the figure of merit to 0.71. The modified experimental map obtained by RESOLVE was readily interpretable, and 42% of the protein chains were traced automatically. Moreover, several rounds of manual fitting and re-fitting were performed using the program COOT [31]. Initially, the model was refined with CNS [32], using a maximum likelihood target that included amplitude and phase probability distributions with NCS restraints. In the later stage of the refinement cycles, the refinement was performed using REFMAC5 [33], incorporated in the CCP 4 package [34]. The final refined model of the dimer in the asymmetric unit contains 360 residues, one MES molecule, two SO4 2- molecules, and 108 water molecules, with a final R work of 19.8% and an R free of 27.3% at 2.3 Å resolution. The first three N-terminal residues of the two chains are missing in the final structure. The side chain atoms of Mse4, Arg12, Arg13, Tyr65, Leu66 and Lys110 in chain A, and Mse4, Arg12, Arg13, Tyr65 and Lys110 in chain B are also missing in the final structure. The stereochemistry of the complex structure is good, as assessed with MOLPROBITY [35, 36]. The Ramachandran plot analysis of this structure revealed that 99.4% of all residues were in the allowed region. The outlier residues are Thr109 and Asn174 in chain A. The electron densities were clear within these disallowed regions. The refinement statistics are summarized in Table 3.
PDB Reference
The structural coordinates of MJ0391 have been deposited in the RCSB (PDB code: 2YXD).
Abbreviations
- NCS:
-
Non-crystallography symmetry
- MAD:
-
Multiple anomalous dispersion
- r.m.s.d.:
-
Root mean square deviation
- CobLc:
-
C-terminal region of CobL.
References
Warren MJ, Raux E, Schubert HL, Escalante-Semerena JC: The biosynthesis of adenosylcobalamin (vitamin B 12 ). Nat Prod Rep 2002, 19: 390–412. 10.1039/b108967f
Scott AI: Discovering nature’s diverse pathways to vitamin B 12 : a 35-year odyssey. J Org Chem 2003, 68: 2529–2539. 10.1021/jo020728t
Roth JR, Lawrence JG, Bobik TA: Cobalamin (coenzyme B 12 ): synthesis and biological significance. Annu Rev Microbiol 1996, 50: 137–181. 10.1146/annurev.micro.50.1.137
Blanche F, Thibaut D, Debussche L, Hertle R, Zipfel F, Müller G: Parallels and decisive differences in vitamin B 12 biosynthesis. Angew Chem Int Ed Engl 1993, 32: 1651–1653. 10.1002/anie.199316511
Battersby AR: How nature builds the pigments of life: the conquest of vitamin B 12 . Science 1994, 264: 1551–1557. 10.1126/science.8202709
Blanche F, Cameron B, Crouzet J, Debussche L, Thibaut D, Vulhorgne M, Leeper FJ, Battersby AR: Vitamin B 12 : How the problem of its biosynthesis was solved. Angew Chem Int Ed Engl 1995, 34: 383–411. 10.1002/anie.199503831
Vévodová J, Graham RM, Raux E, Schubert HL, Roper DI, Brindley AA, Scott AI, Rossener CA, Stamford NP, Stroupe ME, et al.: Structure/function studies on a S -adenosyl- L -methionine-dependent uroporphyrinogen III C methyltransferase (SUMT), a key regulatory enzyme of tetrapyrrole biosynthesis. J Mol Biol 2004, 344: 419–433. 10.1016/j.jmb.2004.09.020
Heldt D, Lawrence AD, Lindenmeyer M, Deery E, Heathcote P, Rigby SE, Warren MJ: Aerobic synthesis of vitamin B 12 : ring contraction and cobalt chelation. Biochem Soc Trans 2005, 33: 815–819. 10.1042/BST0330815
Leech HK, Raux E, McLean KJ, Munro AW, Robinson NJ, Borrelly GP, Malten M, Jahn D, Rigby SE, Heathcote P, et al.: Characterization of the cobaltochelatase CbiXL : evidence for a 4Fe-4S center housed within an MXCXXC motif. J Biol Chem 2003, 278: 41900–41907. 10.1074/jbc.M306112200
Roessner CA, Scott AI: Fine-tuning our knowledge of the anaerobic route to cobalamin (vitamin B 12 ). J Bacter 2006, 188: 7331–7334. 10.1128/JB.00918-06
Scott AI, Roessner CA: Recent discoveries in the pathways to cobalamin (coenzyme B 12 ) achieved through chemistry and biology. Pure Appl Chem 2007, 79: 2179–2188. 10.1351/pac200779122179
Raux E, Lanois A, Warren MJ, Rambach A, Thermes C: Cobalamin (vitamin B 12 ) biosynthesis: identification and characterization of a Bacillus megaterium cobI operon. Biochem J 1998, 335: 159–166.
Roessner CA, Huang K, Warren MJ, Raux E, Scott AI:Isolation and characterization of 14 additional genes specifying the anaerobic biosynthesis of cobalamin (vitamin B12) in Propionibacterium freudenreichii (P. shermanii). Microbiology 2002, 148: 1845–1853.
Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, et al.: Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii . Science 1996, 273: 1058–1073. 10.1126/science.273.5278.1058
Roessner CA, Williams HJ, Scott AI: Genetically engineered production of 1-desmethylcobyrinic acid, 1-desmethylcobyrinic acid a, c -diamide, and cobyrinic acid a, c -diamide in Escherichia coli implies a role for CbiD in C-1 methylation in the anaerobic pathway to cobalamin. J Biol Chem 2005, 280: 16748–16753. 10.1074/jbc.M501805200
Santander PJ, Kajiwara Y, Williams HJ, Scott AI: Structural characterization of novel cobalt corrinoids synthesized by enzymes of the vitamin B 12 anaerobic pathway. Bioorg Med Chem 2005, 14: 724–731.
Kajiwara Y, Santander PJ, Rossener CA, Pérez LM, Scott AI: Genetically engineered synthesis and structural characterization of cobalt-precorrin 5A and -5B, two new intermediates on the anaerobic pathway to vitamin B 12 : definition of the roles of the CbiF and CbiG enzymes. J Am Chem Soc 2006, 128: 9971–9978. 10.1021/ja062940a
Xue Y, Wei Z, Li X, Gong W: The crystal structure of putative precorrin isomerase CbiC in cobalamin biosynthesis. J Struct Biol 2006, 153: 307–311. 10.1016/j.jsb.2005.11.011
Schubert HL, Wilson KS, Raux E, Woodcock SC, Warren MJ: The X-ray structure of a cobalamin biosynthetic enzyme, cobalt-precorrin-4 methyltransferase. Nat Struct Biol 1998, 5: 585–592. 10.1038/846
Schubert HL, Raux E, Wilson KS, Warren MJ: Common chelatase design in the branched tetrapyrrole pathways of heme and anaerobic cobalamin synthesis. Biochemistry 1999, 38: 10660–10669. 10.1021/bi9906773
Frank S, Deery E, Brindley AA, Leech HK, Lawrence A, Heathcote P, Schubert HL, Brocklehurst K, Rigby SE, Warren MJ, Pickersgill RW: Elucidation of substrate specificity in the cobalamin (vitamin B 12 ) biosynthetic methyltransferases. Structure and function of the C 20 methyltransferase (CbiL) from Methanothermobacter thermautotrophicus . J Biol Chem 2007, 282: 23957–23969. 10.1074/jbc.M703827200
Keller JP, Smith PM, Benach J, Christendat D, de Titta GT, Hunt JF: The crystal structure of MT0146/CbiT suggests that the putative precorrin-8w decarboxylase is a methyltransferase. Structure 2002, 10: 1475–1487. 10.1016/S0969-2126(02)00876-6
Holm L, Park J: DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16: 566–567. 10.1093/bioinformatics/16.6.566
Deery E, Schroeder S, Lawrence AD, Taylor SL, Seyedarabi A, Waterman J, Wilson KS, Brown D, Geeves MA, Howard MJ, Pickersgill RW, Warren MJ: An enzyme-trap approach allows isolation of intermediates in cobalamin biosynthesis. Nat Chem Biol 2012, 8: 933–940.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22: 4673–4680. 10.1093/nar/22.22.4673
Gouet P, Courcelle E, Stuart DI, Métoz F: ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 1999, 15: 305–308. 10.1093/bioinformatics/15.4.305
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA: Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA 2001, 98: 10037–10041. 10.1073/pnas.181342398
Otwinowski Z, Minor W: Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276: 307–326.
Terwilliger TC, Berendzen J: Automated MAD and MIR structure solution. Acta Crystallogr 1999, D55: 849–861.
Terwilliger TC: Maximum-likelihood density modification. Acta Crystallogr 2000, D56: 965–972.
Emsley P, Cowtan K: Coot: model-building tools for molecular graphics. Acta Crystallogr 2004, D60: 2126–2132.
Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL: Crystallography & NMR system : A new software suite for macromolecular structure determination. Acta Crystallogr 1998, D54: 905–921.
Murshudov GN, Vagin AA, Dodson EJ: Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr 1997, D53: 240–255.
Collaborative Computational Project, Number 4: The CCP 4 suite: Programs for protein crystallography. Acta Crystallogr 1994, D50: 760–763.
Lovell SC, Davis IW, Arendall WB III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC: Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins 2003, 50: 437–450. 10.1002/prot.10286
Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC: MolProbity : all-atom structure validation for macromolecular crystallography. Acta Crystallogr 2010, D66: 12–21.
Acknowledgments
We thank Ms. Yoko Ukita, Drs. Yoshihiro Agari, Hitoshi Iino and Akeo Shinkai for their assistance during sample preparation. We also thank Drs. Svetlana V. Antonyuk and Richard W. Strange for helpful discussions during the data collection. This work was supported in part by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses, and the X-ray Free Electron Laser Priority Strategy Program (to Y.B.), from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. This work was also supported in part by a grant from the Naito Foundation (2011–164 to Y.B.), the Daiichi-Sankyo Foundation of Life Science (12–039 to Y.B.), and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices”.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BP, SY and YB coordinated and designed the research work. BP performed the data processing and crystallography studies, and prepared the manuscript. YB performed the protein production and crystallization, and participated in drafting the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Padmanabhan, B., Yokoyama, S. & Bessho, Y. Crystal structure of putative CbiT from Methanocaldococcus jannaschii: an intermediate enzyme activity in cobalamin (vitamin B12) biosynthesis. BMC Struct Biol 13, 10 (2013). https://doi.org/10.1186/1472-6807-13-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6807-13-10