
Abstract
Background
The influence of introduction history and post-introduction dynamics on genetic diversity and structure has been a major research focus in invasion biology. However, genetic diversity and structure in the invasive range can also be affected by human-mediated processes in the native range prior to species introductions, an aspect often neglected in invasion biology. Here we aim to trace the native provenance of the invasive tree Acacia pycnantha by comparing the genetic diversity and structure between populations in the native Australian range and the invasive range in South Africa. This approach also allowed us to explore how human actions altered genetic structure before and after the introduction of A. pycnantha into South Africa. We hypothesized that extensive movement and replanting in A. pycnantha’s Australian range prior to its introduction to South Africa might result in highly admixed genotypes in the introduced range, comparable genetic diversity in both ranges, and therefore preclude an accurate determination of native provenance(s) of invasive populations.
Results
In the native range Bayesian assignment tests identified three genetic clusters with substantial admixture and could not clearly differentiate previously identified genetic entities, corroborating admixture as a result of replantings within Australia. Assignment tests that included invasive populations from South Africa indicated similar levels of admixture compared to Australian populations and a lack of genetic structure. Invasive populations of A. pycnantha in South Africa are as genetically diverse as native populations, and could not be assigned to particular native range regions.
Conclusions
Our results indicate that the genetic structure of A. pycnantha in Australia has been greatly altered through various planting initiatives. Specifically, there is little geographic structure and high levels of admixture. While numerous introduction history scenarios may explain the levels of admixture observed in South Africa, planting records of A. pycnantha in Australia suggest that populations were probably already admixed before propagules were introduced to South Africa. These findings have important implications for the management of invasive A. pycnantha populations in South Africa, especially for classical biological control, and more broadly, for studies that aim to understand the evolutionary dynamics of the invasion process.
Similar content being viewed by others

Background
The effect of invasion history on genetic diversity has been documented for numerous species e.g. [1–3]. Many successful invasions are characterized by high genetic diversity – the result of multiple introductions or high propagule pressure from a single source [4], but other invasions are founded by genetically bottlenecked populations which harbour only a small proportion of the total genetic diversity found in their native ranges [5]. Intuitively, high genetic diversity is likely to be beneficial to any species introduced into a new environment. Multiple introductions from distinct native source populations can cause an immediate breakdown of natural gene flow barriers, often leading to admixture (e.g. [6]), increased population genetic diversity [6] hybridization [7] and even genetic novelty [8] in the invaded range. High propagule pressure may also simply enhance the likelihood of introducing suitable genotypes to the new environment [9]. However, a dramatic reduction in genetic diversity, and therefore evolutionary potential, need not limit an invasion, as release from natural enemies [10] and competitors [11], broad environmental tolerance [12], and pre-adaptations [13] may contribute to the success of an invasion.
Disentangling the deterministic and stochastic processes that underlie the genetic diversity and structure of successful invaders is problematic [14], but has important implications for the effectiveness of management interventions, particularly biological control (e.g. [15]). While introduction and post-introduction dynamics have important roles, the genetic structure in the native range population can also influence the way in which introduction histories impact on genetic diversity found in the invasive range. For example, for highly structured populations multiple introduction events from a single population may lead to lower overall diversity than a single introduction sourced from numerous structured native range populations [16].
Tree species introduced for forestry represent a particularly interesting case to explore genetic diversity and structure. These species are typically sampled over large parts of the native range and in large numbers prior to introduction in order to maximize genetic diversity, environmental sampling, and thus evolutionary/breeding potential [16]. Extensive breeding programmes in both the native and introduced range might also ‘pre-adapt’ introduced entities to local environmental conditions [16]. Consequently, the selection, introduction, and establishment of forestry species is often associated with traits linked to successful invasions: high propagule pressure, short generation times, high growth rates [9, 17], as well as high adaptability as mediated through high genetic diversity [4, 6, 8, 18, 19].
Given the complex introduction histories often associated with forestry species, elucidating the processes that shape genetic diversity in the invasive ranges requires accurate and detailed introduction records [16]. Such records are available for the introduction of Acacia pycnantha (Benth.) to South Africa [20]. Acacia pycnantha, commonly known as the golden wattle, is native to south eastern Australia and was introduced to South Africa on two separate occasions in 1865 and 1893 as a potential source of tanbark and for dune reclamation purposes [20]. Experimental plantings of A. pycnantha showed the species to be a promising candidate for tanbark production. While the exact size of both introduction events is unknown, the redistribution of ca. 22–29 million seeds sourced within South Africa throughout the coastal regions of the country is documented [20, 21]. Like many other Australian acacias, A. pycnantha is now invasive in parts of South Africa [22].
In its native range, A. pycnantha is structured into two distinct ecotypes (the dryland and wetland forms) which show a propensity for hybridization [23]. South African populations are genetically less diverse than Australian populations and most closely resemble the wetland form from southern Australia [23]. However, revegetation and roadside plantings from cultivated plants have led to established populations of A. pycnantha in southern Australia and this may obscure phylogeographic signatures in the native range. Given the known impacts of cultivation on the genetic makeup of invasive species, including Australian acacias [8], our overall aim was to use population genetics approaches to better understand the native source(s), genetic diversity, structure, and dynamics of invasive A. pycnantha populations in South Africa. Specifically, we asked: 1) What is the genetic structure in the native range of A. pycnantha? 2) How much of the genetic diversity in A. pycnantha’s native range has been introduced to South Africa? 3) Can source populations of A. pycnantha invading South Africa be identified? 4) Does admixture of geographically isolated genotypes from Australia occur in South Africa?
Results
Genetic diversity
There was evidence of null alleles at one locus (Plop 18) in eight of the populations and so this locus was excluded in further analyses. Tests for Hardy Weinberg equilibrium (HWE) carried out on the remaining seven microsatellites showed that 153 out of 168 locus-by-site comparisons met expectations under HWE. Fifteen locus-by-site comparisons showed an excess of homozygotes. Of these, only three populations out of the 24 sampled populations showed more than one locus deviating from HWE. Three loci (As 2.17, Am435 and Plop 4) were out of HWE for at least three sampled populations. However, in these cases homozygote excess was not attributed to the presence of null alleles in the dataset.
Overall, the number of alleles (N A ), unbiased genetic diversity (H S ), and allelic richness (R S ) were not significantly different in the native range (Australia) than in the invasive range (South Africa). Furthermore, populations in the native range were slightly more structured than populations in the invasive range (Table 1). Overall, A. pycnantha showed very little to no inbreeding in both its native and introduced ranges (Tables 1 and 2).
For levels of intra-population genetic diversities, the mean number of alleles was slightly higher in the introduced populations than in the native range (Table 2). Similarly, expected heterozygosity (HE) was slightly higher in the invaded range than in the native range (Table 2). Fewer private alleles (NP) were found in the invaded ranges in South Africa than in the native range (Table 2).
Genetic structure
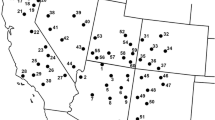
Bayesian assignment analyses in STRUCTURE revealed three different genetic clusters as the optimal number among native range populations while four genetic clusters were identified for the combined native and invasive range datasets (Figure 1 and Additional file 1: Figure S1). For the native range data the three identified genetic clusters only roughly corresponded to three distinct regions from east to west across A. pycnantha’s natural range, including South Australia and Flinders Range (cluster 3, indicated in blue in Figure 1A), the drier parts of Victoria (cluster 2, indicated in yellow in Figure 1A), and New South Wales and some wetter parts of Victoria (cluster 1, indicated in red in Figure 1A). However, the majority of native range populations could not be confidently assigned to any single cluster (q > 0.8), indicative of extensive admixture. Our results also indicate that South African invasive populations probably originated from seeds collected from the entire native range distribution in Australia (Figure 1D). Although the invasive A. pycnantha populations may therefore also represent genetic material from the Flinders Range (dryland form of A. pycnantha, population MEL) it is highly unlikely that this region is a putative source region because of the marked differences observed in leaf morphologies that correspond to distinct ecotypes not found in South Africa [23]. This is supported by chloroplast DNA sequence data obtained from the Flinders Range population that showed no similarity to South African invasive populations [23].

Population genetic structure based on Bayesian assignment tests performed in STRUCTURE. (A) Localities in south eastern (SE) Australia where native Acacia pycnantha populations were sampled. Pie charts indicate overall genotype assignment for each population to particular genetic clusters identified based on native range data only. (B) Localities in South Africa where invasive Acacia pycnantha populations were sampled. (C) Results of the STRUCTURE analysis showing population genetic structure of A. pycnantha populations in its native range (based on native range data only) and (D) combined native (SE Australia) and invasive (South Africa) ranges (combined data). The vertical axes of all STRUCTURE bar plots illustrate the proportional assignment of individual genomes to the inferred genetic groups.
Hierarchical AMOVA of all samples showed almost no differentiation between the native and invasion ranges (1%) but considerable differentiation among populations (13%) whereas the majority of genetic variation resided within populations (86%) (Table 3). The PCoA indicated a close relationship between invasive A. pycnantha from South Africa and native populations from southern Australia, particularly Mt Compass (population MTC) and some Victorian populations (populations HG and NAT) (Figure 2).

Plot of the first two axes of a PCoA showing genetic differentiation based on pairwise Φ PT values for native (Australian) and invasive (South African) populations of Acacia pycnantha .
Discussion and conclusions
We found very low population genetic structure and high levels of gene flow throughout A. pycnantha’s native distribution in south eastern Australia (Figure 1A and C). While this observation supports a previous phylogeographic analysis indicating that admixture frequently occurs in Australian populations, it is also in contrast to the genetic structure previously identified between different ecotypes (dry and wetland forms) of A. pycnantha. These two forms have been estimated to have diverged during the Pleistocene, around 100 KYA [23]. Here we included individuals collected from the same populations as those reported previously on by Ndlovu et al. [23].
Our results make sense as A. pycnantha is known to have been widely moved and planted throughout Australia [24]. Trial plantations have been established at numerous sites in Australia due to the species’ hardiness, drought tolerance and good performance under a range of soil conditions [24]. It has also been widely used for revegetation and soil stabilisation as it shows high natural colonising ability and fast growth under field conditions (http://www.treesforlife.org.au). Like many other wattles, A. pycnantha was heavily harvested in the wild as early as the 1840s. The unsustainable harvesting of wattle bark from natural populations prompted authorities in Victoria to appoint a Wattle Bark Board in 1878 [25]. This Board recommended the establishment of wattle plantations as a sustainable source of tannin bark; these were established in New South Wales, South Australia, Tasmania and Victoria between 1880 and 1900 [26]. For example, the Woods and Forests Department Annual Report (South Australia) for the years 1883–84 [27] mentions that around 40 acres of A. pycnantha plantation were established at Bundaleer, South Australia; during this period testing and dissemination of both Australian native species and alien species was well underway within Australia (D Bush, Australian Tree Seed Centre, CSIRO Plant Industry, personal communication). These practices may have resulted in substantial mixing of genetic material in A. pycnantha’s native range, providing many opportunities for interbreeding to occur between previously allopatric populations [24]. On the other hand, the extensive movement of A. pycnantha within South Africa following introduction may also explain high levels of admixture observed in the invasive range, but it seems more likely that seeds introduced to South Africa in 1893 came from already admixed sources [20].
It is also conceivable that the lack of genetic structure and high levels of admixture observed for A. pycnantha in Australia simply reflects a species with high levels of gene flow. We consider this unlikely, as numerous authors have previously noted that native A. pycnantha populations appear to be structured into two distinct ecotypes [24, 28] and that deep genetic divergence exists between the two [23]. Also, most acacias investigated to date show moderate to high levels of population genetic structure over various spatial scales (e.g. [8, 29, 30]) - including species that co-occur with A. pycnantha in its native range, like A. mearnsii[31] and A. melanoxylon[32].
The role of genetic admixture in successful establishment and invasion has been documented for numerous species [3, 33, 34]. This study is, however, as far as we know, the first to show that anthropogenic actions (plantings for revegetation and the establishment of plantations for bark in this case) may lead to admixture in the native range prior to a species’ introduction into its new range. Of course high levels admixture in South Africa may have also resulted from multiple introductions followed by the extensive redistribution of seed stocks within the country [20, 21]. However, forestry records indicate that the movement and planting of A. pycnantha within Australia predates its introduction to South Africa [26, 27], making a strong case for native range admixture prior to introduction to South Africa. Admixture may enhance the invasion success of a species through pre-adaptation to a wider range of bioclimatic conditions prior to introduction to new regions [35]. Furthermore, because admixture in the native range may increase overall genetic diversity, the likelihood of introducing highly diverse genotypes is enhanced, which also improves the chances of invasion success. Other benefits of admixture to invasion success include masking of deleterious alleles and the creation of novel genotypes [36].
Since the Australian landscape is characterised by many revegetated forests, it is difficult to identify putative sources of invasive A. pycnantha because admixed propagules of A. pycnantha have most likely been introduced to South Africa. However, separate introductions of the biological control agent, the gall-forming wasp Trichilogaster signiventris, from the dryland (Natimuk, Victoria) and the wetland regions of A. pycnantha (Mt Compass) had differential success in South Africa. Such differences in biocontrol efficacy can be indicative of native provenance. It would be useful to test this hypothesis, in line with other work on the influence of phylogeographic structure on the efficacy of biological control (e.g. [15]), as part of the ongoing efforts to improve the contribution of classical biological control to the integrated control of Australian acacias in South Africa [37].
The Acacia pycnantha case demonstrates how human influences on native range population genetic structure prior to a species being even considered for introduction, might ultimately influence evolutionary potential in the introduced range. This is an important but hitherto overlooked dimension of species introduction histories that deserves careful consideration in future studies aimed at better understanding the evolutionary consequences of invasive species.
Methods
Sample collection
Phyllode material of A. pycnantha was collected at seventeen sites throughout the native range (south eastern Australia) and at seven sites from the invaded ranges in South Africa (Table 1). Material was also collected from Mt Compass in South Australia from where the biocontrol agent Trichilogaster signiventris which has successfully established in South Africa was previously collected [38]. For each site, material was collected from between five and 30 trees and preserved in silica gel until DNA extraction. Collection sites were geo-referenced using a handheld GPS.
DNA extraction and PCR conditions
DNA was extracted from phyllode material using a modified [23] CTAB extraction protocol [39]. DNA concentrations were measured using a spectrophotometric nanodrop (Infinite 200 PRO NanoQuant, Tecan Group Ltd, Switzerland) and good quality DNA diluted to 20 ng/μL and stored at −80°C until further use. Eight nuclear microsatellite markers that were previously developed for Acacia mangium[40], A. saligna[41] and Paraserianthes lophantha[42] were cross-amplified in all individuals (see Additional file 2: Table S1). PCR was conducted in two separate multiplexes which were performed using the Qiagen multiplex PCR kit following the manufacturer’s instructions: 5 μL of 2× Qiagen mix, 2 μL of primer mix (containing 2 μM of each primer), 2 μL of RNase free water and 1 μL of DNA template to make up a final volume of 10 μL. The following thermocycle was used: an initial denaturation step of 95°C for 15 min followed by 35 cycles of 94°C for 30 s, an annealing temperature of 57°C for 90s and 72°C for 60 s. A final elongation step of 60°C for 30 min was performed. Separation of PCR fragments was done on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, USA) using GENESCAN TM- 500 (−250) as an internal size standard. Allele scoring was done using GENEMARKER version 1.95 (SoftGenetics LLC, Pennsylvania, USA).
Data analysis
Genetic diversity
Microchecker version 2.2.3 [43] was used to check for null alleles, large allele dropouts and allelic stutter. In addition, FreeNA [44] was also used to examine the presence of null allele frequencies for each locus and population following the expectation maximisation algorithm. All microsatellite loci were tested for departures from the Hardy-Weinberg equilibrium and linkage disequilibrium using the Adegenet package [45] in the R statistical environment [46]. ARLEQUIN version 3.5.1.2 [47] was used to calculate the number of alleles (NA), allelic richness (R S ), observed and expected heterozygosities (HO and HE), fixation indices (FST) and inbreeding coefficients (FIS) for invasive and native ranges and within populations. The mean number of private alleles (N P ) per population was computed in GenAlex v. 6 [48]. Statistical comparisons to evaluate differences in the genetic diversity indices were calculated using permutation procedures as implemented in using FSTAT version 2.9.3.2 [49].
Genetic structure
Bayesian clustering methods implemented in the programme STRUCTURE version 2.3.4 [50] were used to detect the number of genetic clusters (K) present in both the native range and the combined native and invasive range datasets, and to assign individuals probabilistically to these clusters (for K = 1–10). For both analyses, the admixture model with correlated allele frequencies was chosen and 10 replicates of each value of K were run. Each run consisted of a burnin of 10000 MCMC steps, followed by 1000000 iterations. The method of Evanno et al.[51] was used to determine the optimal number of genetic clusters.
To assess the distribution of genotypes in the native and invasive ranges a covariance standardised Principal Co-ordinate Analysis (PCoA) implemented in GenAlex version 6 [48] was used. An analysis of molecular variance (AMOVA) [47] was also performed to partition genetic variation between regions and among regions.
References
Genton BJ, Shykoff JA, Giraud T: High genetic diversity in French invasive populations of common ragweed, Ambrosia artemisiifolia, as a result of multiple sources of introduction. Mol Ecol. 2005, 14: 4275-4285. 10.1111/j.1365-294X.2005.02750.x.
Kelly DW, Muirhead JR, Heath DD, MacIsaac HJ: Contrasting patterns in genetic diversity following multiple invasions of fresh and brackish waters. Mol Ecol. 2006, 15: 3641-3653. 10.1111/j.1365-294X.2006.03012.x.
Kolbe JJ, Glor RE, Rodr L, Lara ADAC, Larson A, Losos JB: Multiple sources, admixture, and genetic variation in introduced anolis lizard populations. Conserv Biol. 2007, 21: 1612-1625. 10.1111/j.1523-1739.2007.00826.x.
Dlugosch KM, Parker IM: Founding events in species invasion: genetic variation, evolution, and the role of multiple introductions. Mol Ecol. 2008, 17: 431-449. 10.1111/j.1365-294X.2007.03538.x.
Bossdorf O, Auge H, Lafuma L, Rogers WE, Siemann E, Prati D: Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia. 2005, 144: 1-11. 10.1007/s00442-005-0070-z.
Lavergne S, Molofsky J: Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc Natl Acad Sci U S A. 2007, 104: 3883-3888. 10.1073/pnas.0607324104.
Gaskin JF, Schaal BA: Hybrid Tamarix widespread in U.S, invasion and undetected in native Asian range. Proc Natl Acad Sci U S A. 2002, 99: 11256-11259. 10.1073/pnas.132403299.
Thompson GD, Bellstedt DU, Byrne M, Millar MA, Richardson DM, Wilson JRU, Le Roux JJ: Cultivation shapes genetic novelty in a globally important invader. Mol Ecol. 2012, 21: 3187-3199. 10.1111/j.1365-294X.2012.05601.x.
Richardson DM, Pyšek P: Plant invasions: merging the concepts of species invasiveness and community invasibility. Prog Phys Geog. 2006, 30: 409-431.
Dewalt SJ, Denslow JS, Ickes K: Natural-enemy release facilitates habitat expansion of the invasive tropical shrub Clidemia hirta. Ecology. 2004, 85: 471-483. 10.1890/02-0728.
Funk JL, Vitousek PM: Resource-use efficiency and plant invasion in low-resource systems. Nature. 2007, 446: 1079-1081. 10.1038/nature05719.
Shea K, Chesson P: Community ecology theory as a framework for biological invasions. Trends Ecol Evol. 2002, 17: 170-176. 10.1016/S0169-5347(02)02495-3.
Schlaepfer DR, Glättli M, Fischer M, Van Kleunen M: A multi-species experiment in their native range indicates pre-adaptation of invasive alien plant species. New Phytol. 2010, 185: 1087-1099. 10.1111/j.1469-8137.2009.03114.x.
Keller SR, Taylor DR: History, chance and adaptation during biological invasion: separating stochastic phenotypic evolution from response to selection. Ecol Lett. 2008, 11: 852-866. 10.1111/j.1461-0248.2008.01188.x.
Goolsby JA, De Barro PJ, Makinson JR, Pemberton RW, Hartley DM, Frohlich DR: Matching the origin of an invasive weed for selection of a herbivore haplotype for a biological control programme. Mol Ecol. 2006, 15: 287-297.
Le Roux JJ, Brown GK, Byrne M, Ndlovu J, Richardson DM, Thompson GD, Wilson JRU: Phylogeographic consequences of different introduction histories of invasive Australian Acacia species and Paraserianthes lophantha (Fabaceae). Divers Distrib. 2011, 17: 861-887. 10.1111/j.1472-4642.2011.00784.x.
Richardson DM, Williams PA, Hobbs RJ: Pine invasions in the southern hemisphere: determinants of spread and invadability. J Biogeogr. 1994, 21: 511-527. 10.2307/2845655.
Okada M, Ahmad R, Jasienuik M: Microsatellite variation points to local landscape plantings as sources of invasive pampas grass (Cortaderia selloana) in California. Mol Ecol. 2007, 16: 4956-4971. 10.1111/j.1365-294X.2007.03568.x.
Williams DA, Muchugu E, Overholt WA, Cuda JP: Colonisation pattern of the invasive Brazilian peppertree Schinus terebinthifolius in Florida. Heredity. 2007, 98: 284-293. 10.1038/sj.hdy.6800936.
Poynton RJ: Tree planting in southern Africa, Vol. 3 other genera. 2009, Pretoria, South Africa: Department of Agriculture, Forestry and Fisheries
Webber BL, Yates CJ, Le Maitre DC, Scott JK, Kriticos DJ, Ota N, McNeill A, Le Roux JJ, Midgley GF: Modelling horses for novel climate courses: insights from projecting potential distributions of native and alien Australian acacias with correlative and mechanistic models. Divers and Distrib. 2011, 17: 978-1000. 10.1111/j.1472-4642.2011.00811.x.
Henderson L: Alien weeds and invasive plants, a complete guide to weeds and invaders in South Africa. PPRI handbooks No. 12, 260. 2001, Pretoria: Plant Protection Research Institute
Ndlovu J, Richardson DM, Wilson JRU, O’Leary M, Le Roux JJ: Elucidating the native sources of an invasive tree species, Acacia pycnantha, reveals unexpected native range diversity and structure. Ann Botany. 2013, 111: 895-904. 10.1093/aob/mct057.
Maslin BR, McDonald MW: Report to the rural industries research and development corporation, publication No. 03/017. Acacia search: evaluation of acacia as a woody crop option for Southern Australia. 2004, Canberra: Joint Venture Agroforestry Program
Victoria, Board of Inquiry: Wattle bark: report of the board of inquiry, together with a statement showing the profit to be derived from the systematic cultivation of wattles. 1892, Melbourne: Government Printer
Frawley J: Joseph Maiden and the National Transitional circulation of wattle Acacia spp. Hist Rec Aust Sci. 2010, 21: 35-54. 10.1071/HR09015.
Brown E: Annual progress report upon state forest administration in South Australia for the year 1883–1884. 1883, Adelaide: Government Printer, 1883-1884.
Costermans L: Native trees and shrubs of south-eastern Australia. 1981, Australia: Rigby
Millar MA, Byrne M, O’Sullivan W: Defining entities in the Acacia saligna (Fabaceae) species complex using a population genetics approach. Aust J Bot. 2011, 59: 137-148. 10.1071/BT10327.
Millar MA, Coates DJ, Byrne M: Genetic connectivity and diversity in inselberg populations of Acacia woodmaniorum, a rare endemic of the Yilgarn Craton banded iron formations. Heredity. 2013, DOI:10.1038/hdy.2013.66
Searle SD, Bell JC, Moran GF: Genetic diversity in natural populations of Acacia mearnsii. Aust J Bot. 2000, 48: 279-286. 10.1071/BT98043.
Playford J, Bell JC, Moran GF: A major disjunction in genetic diversity over the geographic range of Acacia melanoxylon R.Br. Aust J Bot. 1993, 41: 355-368. 10.1071/BT9930355.
Rosenthal DM, Ramakrishnan AP, Cruzan MB: Evidence for multiple sources of invasion and intraspecific hybridization in Brachypodium sylvaticum (Hudson) Beauv. in North America. Mol Ecol. 2008, 17: 4657-4669. 10.1111/j.1365-294X.2008.03844.x.
Kang M, Buckley YM, Lowe AJ: Testing the role of genetic factors across multiple independent invasions of the shrub Scotch broom (Cytisus scoparius). Mol Ecol. 2007, 16: 4662-4673. 10.1111/j.1365-294X.2007.03536.x.
Thompson GD, Robertson MP, Webber BL, Richardson DM, Le Roux JJ, Wilson JRU: Predicting the sub-specific identity of invasive species using distribution models: Acacia saligna as an example. Divers Distrib. 2011, 17: 1001-1014. 10.1111/j.1472-4642.2011.00820.x.
Verhoeven KJF, Macel M, Wolfe LM, Biere A: Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc Biol Sci. 2011, 278: 2-8. 10.1098/rspb.2010.1272.
Wilson JRU, Gairifo C, Gibson MR, Arianoutsou M, Bakar BB, Baret S, Celesti-Grapow L, DiTomaso JM, Dufour-Dror J-M, Kueffer C, Kull CA, Hoffmann JH, Impson FAC, Loope LL, Marchante E, Marchante H, Moore JL, Murphy DJ, Tassin J, Witt A, Zenni RD, Richardson DM: Risk assessment, eradication, and biological control: global efforts to limit Australian acacia invasions. Divers Distrib. 2011, 17: 1030-1046. 10.1111/j.1472-4642.2011.00815.x.
Hoffmann JH, Impson FAC, Moran VC, Donnelly D: Biological control of golden wattle trees (Acacia pycnantha) by a gall wasp Trichilogaster species (Hymenoptera: Pteromalidae) in South Africa. BioControl. 2002, 25: 64-73.
Doyle JJ, Doyle JL: A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem Bull. 1987, 19: 11-15.
Butcher PA, Decroocq S, Gray Y, Moran GF: Development, inheritance and cross-species amplification of microsatellite markers from Acacia mangium. Theor Appl Genet. 2000, 101: 1282-1290. 10.1007/s001220051608.
Millar MA, Byrne M: Characterization of polymorphic microsatellite DNA markers for Acacia saligna (Labill.) H.L.Wendl, (Mimosaceae). Mol Ecol Notes. 2007, 7: 1372-1374. 10.1111/j.1471-8286.2007.01890.x.
Brown GK, Gardner MG: Isolation, characterisation and transferability of microsatellites for Paraserianthes lophantha, Cape Wattle (Leguminoseae: Mimosoideae): a significant weed worldwide. Muelleria. 2011, 29: 87-92.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P: Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004, 4: 535-538. 10.1111/j.1471-8286.2004.00684.x.
Chapuis M-P, Estoup A: Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007, 24: 621-631.
Jombart T: Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008, 24: 1403-1405. 10.1093/bioinformatics/btn129.
R Development Core Team: R: a language and environment for statistical computing. 2009, Vienna: R Foundation for Statistical Computing, http://www.R-project.org,
Excoffier L, Lischer HEL: Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010, 10: 564-567. 10.1111/j.1755-0998.2010.02847.x.
Peakall R, Smouse PE: GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006, 6: 288-295. 10.1111/j.1471-8286.2005.01155.x.
Goudet J: FSTAT version 1.2: a computer programme to calculate F-statistics. J Hered. 1995, 86: 485-486.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005, 14: 2611-2620. 10.1111/j.1365-294X.2005.02553.x.
Acknowledgements
We thank two anonymous reviewers for helpful comments. This work was funded by the DST-NRF Centre of Excellence for Invasion Biology (C•I•B) and the Working for Water Programme through their collaborative research project on “Research for Integrated Management of Invasive Alien Species”. JLR and DMR acknowledge additional funding from the South African National Research Foundation (grant 85417). DMR also received support from the Hans Sigrist Foundation and JLR from the Oppenheimer Memorial Trust. We thank Kate Le Roux for assistance in field collections.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors developed the project and participated in field collections. JLR and JN analysed the data and led the writing of the manuscript. All authors were involved in data interpretation and writing and read, edited, and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Le Roux, J.J., Richardson, D.M., Wilson, J.R. et al. Human usage in the native range may determine future genetic structure of an invasion: insights from Acacia pycnantha. BMC Ecol 13, 37 (2013). https://doi.org/10.1186/1472-6785-13-37
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6785-13-37