
Abstract
Background
Sporadic colorectal cancers (CRC) are multifactorial diseases resulting from the combined effects of numerous genetic, environmental and behavioral risk factors. Genetic association studies have suggested low-penetrance alleles of extremely varied genes to be involved in susceptibility to CRC in Caucasian populations.
Methods
Through a large genetic association study based on 1023 patients with sporadic CRC and 1121 controls, we tested a panel of these low-penetrance alleles to find out whether they could determine "genotypic profiles" at risk for CRC among individuals of the French population. We examined 52 polymorphisms of 35 genes – drawn from inflammation, xenobiotic detoxification, one-carbon, insulin signaling, and DNA repair pathways – for their possible contribution to colorectal carcinogenesis. The risk of cancer associated with these polymorphisms was assessed by calculation of odds ratios (OR) using multivariate analyses and logistic regression.
Results
Whereas all these polymorphisms had previously been found to be associated with CRC risk, especially in Caucasian populations, we were able to replicate the association for only five of them. Three SNPs were shown to increase CRC risk: PTGS1 c.639C>A (p.Gly213Gly), IL8 c.-352T>A, and MTHFR c.1286A>C (p.Ala429Glu). On the contrary, two other SNPs, PLA2G2A c.435+230C>T and PPARG c.1431C>T (p.His477His), were associated with a decrease in CRC risk. Further analyses highlighted genotypic combinations having a greater predisposing effect on CRC (OR 1.97, 95%CI 1.31–2.97, p = 0.0009) than the allelic variants that were examined separately.
Conclusion
The identification of CRC-predisposing combinations, composed of alleles PTGS1 c.639A, PLA2G2A c.435+230C, PPARG c.1431C, IL8 c.-352A, and MTHFR c.1286C, highlights the importance of inflammatory processes in susceptibility to sporadic CRC, as well as a possible crosstalk between inflammation and one-carbon pathways.
Similar content being viewed by others


Background
As in most Western countries, colorectal cancer (CRC) is a major public health issue in France, where it is the second most common cause of death from cancer among adults [1]. Unfortunately, CRC diagnosis is often made at too late a stage and this induces a dismal prognosis, emphasizing the need for prevention and early diagnostic tools [2]. The development of such tools is, nonetheless, highly dependent on the form of cancer being screened. Thus, reliable genetic tests are already available to detect phenotypically well-characterized familial forms of CRC associated with high-penetrance alleles of genes, including APC in familial adenomatous polyposis (FAP), DNA mismatch repair genes in Lynch syndrome or hereditary non-polyposis colorectal cancer (HNPCC), and MUTYH in MUTYH-associated polyposis (MAP) [3]. However, familial diseases together probably make up only a small percentage of all CRCs [2], and the development of comparable screening tests appears more arduous and remote for sporadic cases, which account for the large majority of CRCs.
In contrast with their major role in familial CRCs, heritable factors are not the only components of the etiology of sporadic CRCs. Susceptibility to sporadic CRCs is multifactorial and derives from multiple interactive combinations of numerous low-penetrance alleles and relevant environmental or behavioral risk factors [2, 4]. Independently, each low-penetrance allele contributes modestly to the increase in CRC risk, but its interactions with other susceptibility alleles and environmental factors can lead to a substantial increase in CRC risk, especially when exposed to certain dietary and lifestyle habits [4–6]. The number of interactions is all the higher because the susceptibility genes can be involved in many different biological pathways, which explains the extremely variable phenotype encountered in sporadic CRCs.
Despite much criticism for their non-reproducibility and weak statistical power [6], genetic association studies have been widely used to decypher the mechanisms of cancer susceptibility. Besides, their quality has improved greatly over the past few years [7]. They recently started to produce very valuable results, as illustrated by the identification of several susceptibility loci for colorectal cancers [8–12]. Great expectations can now be held about the results and positive consequences on medical oncology provided by such studies. Beyond the search for susceptibility genes, a global effort is currently being made in the field of sporadic cancers, in order to determine which combinations of genetic variants, i.e., which genetic backgrounds, present at non rare frequencies in the general population, are likely to confer an increase in cancer risk, either alone, or by interacting with usual environmental factors [4, 5, 13, 14].
In order to be part of this effort, we have conducted a case-controlled genetic association study based on a large French population sample. This one already turned out to be very useful by contributing to the finding of the new CRC susceptibility locus at chromosome 8q24.21 made by Zanke et al [9]. Yet, in the present study, our purpose was somehow more modest as we did not attempt to identify new susceptibility loci or variants by a pangenomic approach. Through an exploratory study, we tried to find out whether combinations of variants already known for their possible involvement in carcinogenesis – especially in various Caucasian populations – could determine "genotypic profiles" at risk for CRC among individuals of our French study population. Thus, we focused on 52 allelic variants of 35 candidate genes, selected through a review of the literature on CRC susceptibility, and drawn from five biological pathways relating to inflammation, xenobiotics detoxification, one-carbon, insulin signaling, and DNA repair. Here, we report the results of our investigation on the risk for CRC associated with all 52 allelic variants, analyzed singly or in combinations.
Methods
Experimental design and study population
From December 2002 to March 2006, two groups of 1023 patients (632 males and 391 females; mean ± SD age at diagnosis 65.7 ± 10.1 years) and 1121 controls (609 males and 512 females; mean ± SD age at inclusion 61.9 ± 10. years), all of Caucasian origin, were recruited within the Pays de la Loire region of France.
Details of the study population characteristics have been described elsewhere [15]. In short, all cases were patients recruited in regional hospitals and clinics, with a personal history of colorectal cancer diagnosed at an age of ≥ 40 years old. Any patient suspected of having a familial form of colorectal cancer (either Familial Adenomatous Polyposis, Hereditary Non Polyposis Colorectal Cancer or MUTYH-Associated Polyposis) was excluded from the study. Age- and sex-matched controls were recruited at two regional Health Examination Centres; all were ≥ 40 years old and had no family history of colorectal cancer or polyps. Two venous blood samples were collected from each participant in an anonymous manner, after they had legally provided a written informed consent. The study was approved by both the local ethics committee CCPPRB (Consultative Committee for the Protection of Person in Biomedical Research) and by the national French ethics committee CNIL (National Commission for Data protection and the Liberties). Each participant also answered the same one-page standardized questionnaire pertaining to life and food habit information. Endoscopy and histology reports were obtained for each patient.
Selection of the low-penetrance genes and allelic variants
A review of the literature focused on susceptibility to sporadic colorectal cancer highlighted five biological pathways frequently cited for their role in colorectal carcinogenesis: inflammation, one-carbon, signaling insulin, xenobiotics detoxification, and DNA repair. In these pathways, we selected 35 genes reported as known or possible factors predisposing to CRC (ALOX5, ALOX12, IL6, IL8, PLA2G2A, PDL2, PTGS1, PTGS2, PPARG, CYP1A2, CYP2E1, CYP1B1, CYP2C9, EPHX1, GSTA1, GSTM1, GSTM3, GSTP1, GSTT1, NQO1, SULT1A2, UGT1A1, UGT1A6, GH1, IGF1, IGFBP3, IRS1, VDR, CBS, MTHFD1, MTHFR, MTR, MTRR, TYMS, OGG1), and chose to investigate 52 polymorphisms -46 single nucleotide polymorphisms (SNPs) and 6 complex polymorphisms – in these 35 genes (Table 1). Among these 52 polymorphisms, 38 polymorphisms were exclusively selected through our review of the literature, because of their possible involvement in the predisposition to CRC [16–30]; most of these polymorphisms reportedly modified the in vitro activity of the corresponding gene. It should be noted that 6 of these polymorphisms – found in CYP1A2, CYP2E1, CYP1B1 and CYP2C9-, which were tested previously in our study population [15], are still cited in the present study, given that they were included in the combined analyses described below. The remaining 14 SNPs, located in the candidate genes PLA2G2A, PTGS1, PTGS2, and PPARG, were first found by a dHPLC screening in two samples of 50 patients and 50 controls randomly chosen within the whole study population. In fact, we chose to select these 14 SNPs, because of their uneven distribution between the two samples. With the exception of the 4 SNPs in PLA2G2A, the other ten SNPs were also found by our bibliographical survey on susceptibility to CRC [5, 17, 31–34].
As regards the nomenclature that we used for the SNPs, we referred to the databases dbNSP, SNPPER and SNP500 Cancer for the ID numbers, and we followed the Human Variation Society (HGVS) mutation nomenclature for the description of nucleotide and proteic sequence variants [35]. Thereby, we corrected wrong notations abundantly used in literature for some of the selected SNPs (e.g., PPARG C161T instead of c.1431C>T, or MTHFR A1298C instead of c.1286A>C).
Genotype analysis
Every study participant was genotyped for the 52 polymorphisms selected. Genotypes were determined using high-throughput TaqMan allelic discrimination tests for the 46 SNPs, and fluorescent multiplex PCRs for the 6 complex polymorphisms [for details, see Additional files 1 and 2].
Statistical analysis
Single-SNP association study
All polymorphisms were tested for Hardy-Weinberg equilibrium by a χ2 test in both patients and controls. An appraisal was made on the association between allelic variants and CRC risk, by calculation the odds ratio (OR) of cancer cases together with 95 percent confidence intervals (CI). Association tests consisted of comparing genotype heterogeneity between the groups of patients and controls. For each allelic variant, homozygotes for the major allele were used as the reference group and compared to the other two groups of heterozygotes and homozygotes for the minor allele considered either separately (codominant model), or gathered in a same group (dominant model). We used logistic regression models for assessing single SNP effects adjusted for gender and age covariates. To estimate the risk for CRC, we first performed multivariate analyses based on unconditional logistic regression, adjusting for sex and age at date of reference (date of diagnosis for patients, and date of blood sample collection for controls). To detect a potential overestimation of the odds ratio by unconditional logistic regression, we then employed conditional logistic regression on 811 age- and sex-matched pairs of individuals (75.6% of the whole study population, including 2*324 women, and 2*487 men). In each case, the significance of ORs was assessed by calculation of p-values derived from likelihood-ratio tests.
Multiple testing of 52 polymorphisms implies inevitably a certain rate of false positive results. A correction is therefore required to lower as much as possible the rate of false positives, even though the need to adjust multiple comparisons has been questioned by Rothman [36]. Yet, classical methods of correction such as Bonferroni's adjustment are too drastic for genetic association studies and lead to the rejection of many results, including true associations. We therefore followed the approach proposed by Storey et al., and we estimated the false discovery rate (FDR), more appropriate in large sets of hypotheses than multiple testing procedures [37]. For this estimation, we calculated the q-value, setting the threshold at 0.05. We considered findings that met a false discovery rate (FDR) criterion of 20% to be robust.
Multiple-SNP association study
Modeling strategy for analysis of genotypic combinations
According to the results of single polymorphisms analyses, we tested all possible combinations of N polymorphisms composed by the N allelic variants found to be significantly associated with CRC risk, and by polymorphisms associated with marginally or non-significant ORs having a p value comprised between 0.05 and 0.25. As a comparison, we also analyzed a hundred of combinations of N polymorphisms randomly selected from any of the 52 ones included in our study. For each polymorphism, the genotype was coded 0 if the rare variant was absent, 1 if present at heterozygous state, 2 if present at homozygous state. Because most of the homozygotes for the minor allele of the polymorphisms tested were often extremely rare, we assumed in our model that the rare effect allele was dominant. In other terms, if a polymorphism predisposed to CRC, genotypes 1 and 2 were considered predisposing to CRC; on the contrary, genotypes 1 and 2 were considered protective if the effect of the minor variant of the polymorphism was rather protective. For each polymorphism included in a combination, we therefore tested two hypotheses: 1. either we considered that the minor allele was predisposing to CRC and therefore the CRC-predisposing genotypes were 1 and 2, whereas genotype 0 was considered protective; 2. or we considered that the minor allele was associated with a decreased CRC risk, and therefore 0 was considered to be the predisposing genotype, whereas genotypes 1 and 2 were considered protective. Thus, for every combination of N SNPs tested, we also tested every possible combination of x protective and N-x predisposing minor alleles (with x ranging from 0 to N), each combination tested representing a different model.
For each test of a combination or model, we compared three groups. The first group was composed by individuals exhibiting genotype 0 for the N SNPs, i.e., carrying the frequent allele at homozygous state for the N SNPs; this group, theoretically the most frequent group in the study cohort, was set as a reference, and the relating combination of genotypes was considered as the "reference pattern" exhibiting a null or very weak effect on CRC risk. The second group was composed of individuals exhibiting patterns of genotypic combinations composed of N predisposing genotypes (predisposing patterns). The third group comprised the individuals exhibiting all other possible combinations of genotypes (mixed patterns).
In the same way as for single SNP association study, heterogeneity between the groups of patients and controls was translated into colorectal cancer risk associated with genotypic patterns, assessed by OR calculation together with 95 percent confidence intervals. We first used unconditional logistic regression, adjusting for sex and age, and we compared the results observed to those obtained by conditional logistic regression analysis restricted to 811 age- and sex-matched pairs of individuals. We eventually assessed the robustness of the associations observed by applying the false discovery rate (FDR) method, setting the threshold at 0.05.
Validation of the logistic regression model
In order to validate the logistic regression model, we applied a standard Monte Carlo bootstrap (random resampling with replacement from the original dataset) procedure based on 1000 replicates. We calculated the 5th and 95th percentiles from the resulting distributions to determine the lower and upper limits of the confidence interval.
Alternatively, we examined the internal consistency of the results obtained for the model. Analyses of genotypic combinations were replicated on stratifications of the study population determined according to age, geographical origin, random selection, or inference of population structure [Additional file 1].
Results
The allele frequencies we found for each of the 52 polymorphisms tested were consistent with those reported in literature and in dbSNP http://www.ncbi.nlm.nih.gov/projects/SNP/ for Caucasian populations. Two of the 52 polymorphisms analyzed turned out to be monomorphic. The 50 others were all at Hardy-Weinberg equilibrium. Results of the unconditional logistic regression analyses for CRC association with polymorphisms considered independently are described in Table 1. Alternative results of conditional logistic regression restricted to 811 age- and sex-matched pairs of individuals are reported in Additional file 3.
Six associations between CRC risk and allelic variants were determined by both unconditional and conditional logistic regression analyses. For SNPs PTGS1 c.639C>A (p.Gly213Gly), IL8 c.-352T>A, and MTHFR c.1286A>C (p.Ala429Glu), minor alleles appeared associated with an increase in CRC, whereas for SNPs PLA2G2A c.435+230C>T, PPARG c.1431C>T (p.His477His), they were associated with a decrease in CRC risk. Application of the false discovery rate to the results obtained by unconditional logistic regression analysis showed a value of about 0.5 for these five findings, suggesting that the associations were not very robust. Yet, on the contrary, q-values determined from conditional logistic regression analysis were found between 0.07 and 0.26 for the same findings, rather suggesting quite robust associations [Additional file 3]. Our analyses also showed a protective effect of the variant GSTM1 null carried at the heterozygous state on CRC risk. However, this observation is not relevant at the biological level since the loss of activity of the GSTM1 enzyme, or GSTM1 deficiency, is associated with a deletion carried on both alleles. In fact, complementary analyses in our study population revealed that individuals carrying two deleted alleles did not have a significantly different risk of CRC compared to carriers of one or zero deleted alleles (OR 1.08, 95% CI 0.91–1.21, p = 0.4), which is consistent with two meta-analyses performed on this polymorphism [19, 38]. Therefore, the CRC-predisposing effect observed for GSTM1 null allele is to consider with great caution, all the more because the q-value of 0.4 calculated for this finding would rather suggest a false positive result. Two additional CRC-predisposing effects were observed for SNPs PLA2G2A c.-859C>G and CYP1B1 c.1294C>G by conditional logistic regression analysis [Additional file 3], but were not found when using unconditional method applied to the whole study population; they were therefore considered as false positives.
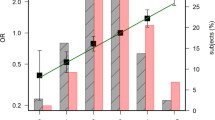
Since five SNPs were undoubtfully found associated with a modification of CRC risk in single-SNP analyses, we focused our further analyses on combinations of five allelic variants, as described in Methods. Among all the genotypic combinations tested, only one showed a statistically significant association with an increased risk of CRC, which appeared robust according to the calculation of FDR (q-value < 0.1). This combination was composed by the very same five SNPs that had been found independently associated with CRC risk from single-SNP analyses. Additional file 4 uses the example of this precise set of five SNPs to illustrate the different models of genotypic combinations tested for every set of five polymorphisms – picked from the 52 polymorphisms of the study – which we tested. The most predisposing patterns for this combination, presented in Table 2, combine genotypes 1 and 2 for PTGS1 c.639C>A, IL8 c.-352T>A, and MTHFR c.1286A>C, and 0 for PLA2G2A c.435+230C>T and PPARG c.1431C>T. These predisposing patterns appeared to be associated with a highly significant increase in colorectal cancer, either compared to the reference pattern alone (OR 2.65; 95% CI 1.58–4.42 with p = 0.0005), or compared to reference and mixed patterns gathered (OR 1.97; 95% CI 1.31–2.97 with p = 0.0009).
Validation of the model by bootstrapping and examination of internal consistency strengthened the above results. By bootstrapping, we calculated a mean ± SD OR of 1.86 ± 0.41 (95%CI 1.23–2.85, p = 0.003). Examination of internal consistency confirmed a trend in an increased CRC risk associated with the predisposing combination patterns, regardless of the mode of stratification used [Additional file 5]. No effect of gender or anatomical sub-location was noted. Most of the associations observed were statistically significant, but certain stratifications determined according to geographical origin and/or age displayed a greater effect regarding the predisposing combination patterns (Table 3). Among the 2144 individuals composing the whole study population, the strongest association was found in individuals of ≤ 67 years of age and originating from the French département of the Vendée.
Discussion
In this study, we have used a candidate gene approach to examine the associations between colorectal cancer risk and 52 allelic variants distributed in 35 genes drawn from pathways of inflammation, metabolism of xenobiotics detoxification, one-carbon, insulin signaling, and DNA repair. To our knowledge, we are describing here within one of the most comprehensive investigations on populations of this kind, covering more than 1000 patients with sporadic colorectal cancer, and 1000 controls, i.e., in the range of 500–2000 case-control pairs defined by Brennan to detect the statistically significant effect of polymorphisms [39]. Obviously, the list of polymorphisms which we designed here is non-comprehensive, and it must be seen as a panel test, a first attempt in the search for CRC-predisposing "genetic profiles".
With the exception of the 4 SNPs in PLA2G2A that we had selected by dHPLC, all the allelic variants chosen for the present study had previously been found to be associated with a modification of CRC risk in at least one study. However, we have been able to replicate the association with CRC risk for only 5 of the 52 polymorphisms tested (Table 1). By independent single-SNP analyses, three SNPs were shown to increase CRC risk: PTGS1 c.639C>A, IL8 c.-352T>A, and MTHFR c.1286A>C. Two other SNPs, PLA2G2A c.435+230C>T and PPARG c.1431C>T, were on the contrary associated with a decrease in CRC risk. Combinations of the CRC-predisposing alleles relating to these five variants determine "genotypic profiles" at significantly higher risk of CRC (OR 1.97, 95% CI 1.31–2.97). Among the individuals exhibiting these profiles, younger individuals (≤ 67 years) from the Vendée seem to be even more predisposed to CRC (OR 6.36, 95% CI 1.44–28.09). However, the size of the population sample analyzed here is too small to draw definitive conclusions on any age- or geographical-effect. In the same way, the consistency of the risk profile was lost in some of the study population subgroups used for the analyses reported in Additional file 5.b, certainly because of their relatively small size. Caution is therefore required, when considering the true significance of this risk profile, even though bootstrapping results tend to show its robustness [Additional file 5.a]. It is noteworthy that a sixth polymorphism, GSTM1 null allele, showed a protective effect in independent analyses of allelic variants, but the significance of its association with CRC risk remained dubious, and was not confirmed by multiple-SNP analyses, contrary to the five other polymorphisms.
To investigate further the CRC-predisposing combinations that emerged from our analyses, we tested their possible interactions with the environmental and lifestyle risk factors reported in our study questionnaire (physical activity, cooking methods, and consumption of alcohol, tobacco, red meat, cold cuts, white meat and poultry bread, dairy products, fish, fruits, pastries, or vegetables), by use of SNPStats [40]. Indeed, in the same study population, we had previously observed an interaction between allelic combinations of CYP genes and consumption of red meat which leads to a strong increase in CRC risk [15]. In the present study, we did not observe any comparable gene-environment interaction that could remain statistically significant throughout multiple adjustment and/or test for internal consistency in patient and control sub-groups (data not shown). Given that four of the SNPs composing the CRC-predisposing combinations are related to inflammation, the most relevant "environmental" factor to be tested here would have actually been NSAIDs treatment. But, since this item did not figure in our questionnaire on life-habits [15] – we assumed that it would have introduced a bias in the design of the control group-, we were unfortunately unable to analyze its interaction with the CRC-predisposing genotypic combinations of the five SNPs mentioned above.
The respective biological impact of the five variants PTGS1 c.639C>A, IL8 c.-352T>A, MTHFR c.1286A>C, PLA2G2A c.435+230C>T, and PPARG c.1431C>T provide some clues to the predisposition to CRC associated with some of the genetic combinations that they compose. Thus, PLA2G2A c.435+230C>T and PTGS1 c.639C>A, belong to genes which encode proteins following each other in the enzymatic cascade of the arachidonic acid pathway. PLA2G2A catalyzes the hydrolysis of membrane phosopholipids, thereby releasing unsaturated fatty acids, including arachidonic acid [41]. The latter becomes the substrate of PTGS1, which in turn catalyzes the formation of prostaglandin H2 (PGH2), a precursor for a number of inflammatory molecules – eicosanoids – that promote colorectal carcinogenesis [42]. Until now, out of the potential key players in the CRC process, more attention has been given to another element of the arachidonic acid pathway, PTGS2, rather than to PTGS1 or PLA2G2A [31, 43]. The interest for PTGS2 in CRC risk stems from its expression induced by pathophysiological conditions such as tumorigenesis or inflammatory situations [16]. However, we found no significant modification of CRC risk associated with any of the four PTGS2 SNPs we tested (Table 1). In contrast, PTGS1 is constitutively expressed and its implication in CRC risk has not been investigated very much, since its carcinogenetic role through induction of PTGS2 has been suggested only recently [16, 34]. Thus, to our knowledge, the synonymous polymorphism c.639C>A (p.Gly213Gly) that we found as having a predisposing effect on CRC (OR 1.24) at the heterozygous (CA) or homozygous state (AA), had not been examined within this context to date and its precise impact on PTGS1 activity would require further functional studies. In the same way, the four PLA2G2A SNPs we studied have not yet been tested within the CRC risk context. Therefore, there is no possible point of comparison for the protective effect relating to CRC that we found for the allele c.435+230T carried at homozygous state (OR 0.80, 95%CI 0.66–0.98). Because of its localization within the Mom-1 (Min modifier) locus, and its promotion of tumors in APCMin mice, it had first been suggested that PLA2G2A may represent a tumor suppressor gene involved in familial forms of CRC [44]. As a result, such a role of PLA2G2A in FAP genesis had been ruled out in humans [41, 45]. However, the contribution of PLA2G2A to sporadic CRC predisposition cannot be excluded, and it has been suggested that the increase in PLA2G2A expression could cause the accumulation of arachidonic acid, a molecule likely to have pro-apoptotic properties [41]. A hypothesis for the protective role of variant c.435+230T might therefore be its enhancement of PLA2G2A expression.
PPARG c.1431C>T (aka C161T) and IL8 c.-352T>A, also relate to inflammation or immune response. PPARγ (peroxisome proliferator-activated receptor gamma) is a nuclear receptor tightly linked to the arachidonic acid pathway, in that it is activated by various eicosanoids [46]. Essential to adipocyte differentiation and to regulation of lipid metabolism, PPARγ is thought to have overall tumor suppressive properties, exerted notably in CRC [46–48]. A protective effect on CRC risk had been reported for the rare allele of variant PPARG c.36C>G (p.Pro12Ala) [49, 50], but we were not able to reproduce this observation in our study population, perhaps because the reference studies were designed on smaller populations – about 200 case-control pairs-. On the other hand, we noted a significantly decreased risk of CRC associated with the rare allele of variant c.1431C>T (p.His477His) at the homozygous state (OR 0.30, 95%CI 0.14–0.65). These results are consistent with those reported in an earlier study on colorectal adenoma [48], but they are inconsistent with the contrary observations made by another team investigating CRC [33]. Given the controversial effect assigned to PPARG c.1431C>T, functional assays would be required to understand the exact role of this little-investigated polymorphism.
As regards IL8, it encodes a pro-inflammatory chemokine, released by infiltrating lymphocytes, in response to exposure of the colonic epithelium to toxic and pathologic challenges [50]. The allelic variant IL8 c.-352T>A has been found to be associated with CRC risk, even though the effect attributed to rare allele A goes from null [51], to predisposing or even protective [17, 52]. As regards our study, we found that genotypes c.352AA or c.352TA were associated with an elevated risk of CRC compared to genotype c.352TT (OR 1.21, 95%CI 1.01–1.46). The debate on the true carcinogenetic role of variant c.-352T>A probably comes from its controversial impact on IL8 transcription, sometimes described as an enhancement [51], sometimes as a downregulation [52]. As IL8 contributes to chronic inflammation, it would make sense that its overexpression would increase the risk for CRC, and therefore the allele c.-352A may rather enhance the transcriptional activity of the gene.
The fifth gene highlighted by our analyses, MTHFR (5,10-methylenetetrahydrofolate reductase), is part of the one-carbon metabolic pathway, involved in both DNA methylation and DNA synthesis. The MTHFR enzyme synthesizes 5-methyltetrahydrofolate, the primary circulatory form of folate, which represents a fundamental methyl donor in cellular metabolism [26]. The influence of MTHFR activity on folate status could be important in CRC neoplasia, since folate deficiency could cause DNA hypomethylation, and/or induce uracil misincorporation during DNA synthesis, which would lead to mutations and chromosomal damage [26, 27, 53]. Two polymorphisms c.665C>T (also reported as C677T) and c.1286A>C (aka c.1298A>C), have been widely tested for modification of CRC risk, because of the reduction in MTHFR activity induced by their minor allele. In the present study, we found no effect for c.665C>T, whereas we observed an elevated risk of CRC associated with genotypes c.1286CC or c.1286AC compared to genotype c.1286AA (OR 1.21, 95%CI 1.02–1.44). At first sight, these results appear difficult to compare with the greatly inconsistent or even conflicting earlier results, and reviewed in two recent comprehensive meta-analyses [54, 55]. However, this discrepancy in the overall results could be explained by the variation of the polymorphisms effect according to folate status. Mu et al. suggested indeed that genotypes reducing MTHFR activity – here c.1286CC or c.1286AC – would favor cancer risk when dietary folate levels are low, by inducing a DNA hypomethylation causing DNA damage and mutations [56]. When folate levels are adequate, the reduced MTHFR activity induced by the same genotypes would lead to a great pool of methylenetetrahydrofolate available for DNA synthesis, and therefore prevent cancer by diminishing uracil misincorporation. Moreover, it has been suggested that, according to its administration prior or further to the existence of preneoplastic lesions, folate would rather prevent or increase tumor development, respectively [53].
All these biological data considered, our work underlines the important contribution of inflammatory processes to CRC susceptibility in our study population, and it points to a possible crosstalk between inflammation and one-carbon pathways. The four allelic variants of genes PTGS1, PLA2G2A, PPARG, and IL8 might favor inflammatory processes, whereas the MTHFR allelic variant could induce a DNA hypomethylation altering the expression of the putative tumor suppressor gene PPARG. Two recent studies on diabetes and cardiovascular diseases have suggested a common pathobiological mechanism between the inflammation process and genotype TT of MTHFR c.665C>T (aka C677T), i.e., a genotype inducing a lower MTHFR activity like genotype MTHFR c.1286CC [57, 58]. However, our results do not enable to conclude to an interaction between the five allelic variants. Indeed, the odd ratios associated with the observed CRC-predisposing combinations are certainly not strong enough, and our method is not the most appropriate one for an investigation on gene-gene interactions.
Conclusion
Combinations of alleles PTGS1 c.639A, PLA2G2A c.435+230C, PPARG c.1431C, IL8 c.-352A, and MTHFR c.1286C determine "genotypic profiles" at significantly higher risk of CRC (OR 1.97, 95% CI 1.31–2.97), in a small percentage of the French population. The identification of these CRC-predisposing combinations highlights the importance of inflammatory processes in susceptibility to sporadic CRC, as well as a possible crosstalk between inflammation and one-carbon pathways.
To find their significance, our results would obviously need to be reproduced. Indeed, replication represents a key step for establishing the credibility of any genotype-phenotype association [59]. Only a validation in other Caucasian populations would therefore enable to consider the five above variants and their related genotypic combinations as relevant risk markers for sporadic CRC. Moreover, even though the size of our population sample is quite large, it is not large enough to draw definitive conclusions on the statistical significance of the polymorphisms-disease associations observed. In order to reach such conclusions, our results should be combined to a meta-analysis, since this tool is particularly robust to estimate population-wide effect of genetic risk factors in diseases [60]. In any case, our observations would warrant investigations on possible interactions between NSAIDs and combinations of polymorphisms from the five genes we have highlighted here, which our questionnaire unfortunately did not enable us to achieve. The results of our study also call for more comprehensive investigations into polymorphisms of inflammation genes – especially those belonging to the arachidonic acid pathway-, as candidate risk factors for CRC. In fact, the CRC-predisposing combinations we identified account for only 5% of our study population, which means that many more predisposing combinations, involving different and maybe common polymorphisms, are still to be found. Very large genetic association studies on candidate genes of inflammation may make it possible to find the most frequent predisposing combinations, and thus make the first step towards drawing up recommendations for clinical practice guidelines regarding sporadic CRC.
References
Remontet L, Esteve J, Bouvier AM, Grosclaude P, Launoy G, Menegoz F, Exbrayat C, Tretare B, Carli PM, Guizard AV, Troussard X, Bercelli P, Colonna M, Halna JM, Hedelin G, Mace-Lesec'h J, Peng J, Buemi A, Velten M, Jougla E, Arveux P, Le Bodic L, Michel E, Sauvage M, Schvartz C, Faivre J: Cancer incidence and mortality in France over the period 1978–2000. Rev Epidemiol Sante Publique. 2003, 51 (1 Pt 1): 3-30.
de la Chapelle A: Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004, 4 (10): 769-780. 10.1038/nrc1453.
Davidson NO: Genetic testing in colorectal cancer: who, when, how and why. Keio J Med. 2007, 56 (1): 14-20. 10.2302/kjm.56.14.
Hunter DJ, Riboli E, Haiman CA, Albanes D, Altshuler D, Chanock SJ, Haynes RB, Henderson BE, Kaaks R, Stram DO, Thomas G, Thun MJ, Blanche H, Buring JE, Burtt NP, Calle EE, Cann H, Canzian F, Chen YC, Colditz GA, Cox DG, Dunning AM, Feigelson HS, Freedman ML, Gaziano JM, Giovannucci E, Hankinson SE, Hirschhorn JN, Hoover RN, Key T, et al: A candidate gene approach to searching for low-penetrance breast and prostate cancer genes. Nat Rev Cancer. 2005, 5 (12): 977-985. 10.1038/nrc1754.
Goodman JE, Mechanic LE, Luke BT, Ambs S, Chanock S, Harris CC: Exploring SNP-SNP interactions and colon cancer risk using polymorphism interaction analysis. Int J Cancer. 2006, 118 (7): 1790-1797. 10.1002/ijc.21523.
Tabor HK, Risch NJ, Myers RM: Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet. 2002, 3 (5): 391-397. 10.1038/nrg796.
Naccarati A, Pardini B, Hemminki K, Vodicka P: Sporadic colorectal cancer and individual susceptibility: a review of the association studies investigating the role of DNA repair genetic polymorphisms. Mutat Res. 2007, 635 (2–3): 118-145.
Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, et al: A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007, 39 (8): 984-988. 10.1038/ng2085.
Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bézieau S, O'Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous ME, Campbell H, et al: Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007, 39 (8): 989-994. 10.1038/ng2089.
Picelli S, Vandrovcova J, Jones S, Djureinovic T, Skoglund J, Zhou XL, Velculescu VE, Vogelstein B, Lindblom A: Genome-wide linkage scan for colorectal cancer susceptibility genes supports linkage to chromosome 3q. BMC Cancer. 2008, 8 (1): 87-10.1186/1471-2407-8-87.
Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E, Cetnarskyj R, Cartwright N, Semple C, Clark AJ, Reid FJ, Smith LA, Kavoussanakis K, Koessler T, Pharoah PD, Buch S, Schafmayer C, Tepel J, Schreiber S, Volzke H, Schmidt CO, Hampe J, Chang-Claude J, Hoffmeister M, Brenner H, Wilkening S, Canzian F, Capella G, et al: Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008, 40 (5): 631-637. 10.1038/ng.133.
Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A, Sullivan K, Jaeger E, Fielding S, Rowan A, Vijayakrishnan J, Domingo E, Chandler I, Kemp Z, Qureshi M, Farrington SM, Tenesa A, Prendergast JG, Barnetson RA, Penegar S, Barclay E, Wood W, Martin L, Gorman M, Thomas H, Peto J, Bishop DT, et al: A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008, 40 (5): 623-630. 10.1038/ng.111.
Onay VU, Briollais L, Knight JA, Shi E, Wang Y, Wells S, Li H, Rajendram I, Andrulis IL, Ozcelik H: SNP-SNP interactions in breast cancer susceptibility. BMC Cancer. 2006, 6: 114-10.1186/1471-2407-6-114.
Wu X, Gu J, Grossman HB, Amos CI, Etzel C, Huang M, Zhang Q, Millikan RE, Lerner S, Dinney CP, Spitz MR: Bladder cancer predisposition: a multigenic approach to DNA-repair and cell-cycle-control genes. Am J Hum Genet. 2006, 78 (3): 464-479. 10.1086/500848.
Küry S, Buecher B, Robiou-du-Pont S, Scoul C, Sebille V, Colman H, Le Houérou C, Le Neel T, Bourdon J, Faroux R, Ollivry J, Lafraise B, Chupin LD, Bézieau S: Combinations of cytochrome P450 gene polymorphisms enhancing the risk for sporadic colorectal cancer related to red meat consumption. Cancer Epidemiol Biomarkers Prev. 2007, 16 (7): 1460-1467. 10.1158/1055-9965.EPI-07-0236.
Goodman JE, Bowman ED, Chanock SJ, Alberg AJ, Harris CC: Arachidonate lipoxygenase (ALOX) and cyclooxygenase (COX) polymorphisms and colon cancer risk. Carcinogenesis. 2004, 25 (12): 2467-2472. 10.1093/carcin/bgh260.
Landi S, Moreno V, Gioia-Patricola L, Guino E, Navarro M, de Oca J, Capella G, Canzian F: Association of common polymorphisms in inflammatory genes interleukin (IL)6, IL8, tumor necrosis factor alpha, NFKB1, and peroxisome proliferator-activated receptor gamma with colorectal cancer. Cancer Res. 2003, 63 (13): 3560-3566.
Yamada Y, Hamajima N, Kato T, Iwata H, Yamamura Y, Shinoda M, Suyama M, Mitsudomi T, Tajima K, Kusakabe S, Yoshida H, Banno Y, Akao Y, Tanaka M, Nozawa Y: Association of a polymorphism of the phospholipase D2 gene with the prevalence of colorectal cancer. J Mol Med. 2003, 81 (2): 126-131.
de Jong MM, Nolte IM, te Meerman GJ, Graaf van der WT, de Vries EG, Sijmons RH, Hofstra RM, Kleibeuker JH: Low-penetrance genes and their involvement in colorectal cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 2002, 11 (11): 1332-1352.
Landi S, Gemignani F, Moreno V, Gioia-Patricola L, Chabrier A, Guino E, Navarro M, de Oca J, Capella G, Canzian F: A comprehensive analysis of phase I and phase II metabolism gene polymorphisms and risk of colorectal cancer. Pharmacogenet Genomics. 2005, 15 (8): 535-546.
Sachse C, Smith G, Wilkie MJ, Barrett JH, Waxman R, Sullivan F, Forman D, Bishop DT, Wolf CR: A pharmacogenetic study to investigate the role of dietary carcinogens in the etiology of colorectal cancer. Carcinogenesis. 2002, 23 (11): 1839-1849. 10.1093/carcin/23.11.1839.
Logt van der EM, Bergevoet SM, Roelofs HM, van Hooijdonk Z, Te Morsche RH, Wobbes T, de Kok JB, Nagengast FM, Peters WH: Genetic polymorphisms in UDP-glucuronosyltransferases and glutathione S-transferases and colorectal cancer risk. Carcinogenesis. 2004, 25 (12): 2407-2415. 10.1093/carcin/bgh251.
Innocenti F, Grimsley C, Das S, Ramirez J, Cheng C, Kuttab-Boulos H, Ratain MJ, Di Rienzo A: Haplotype structure of the UDP-glucuronosyltransferase 1A1 promoter in different ethnic groups. Pharmacogenetics. 2002, 12 (9): 725-733. 10.1097/00008571-200212000-00006.
Le Marchand L, Donlon T, Seifried A, Kaaks R, Rinaldi S, Wilkens LR: Association of a common polymorphism in the human GH1 gene with colorectal neoplasia. J Natl Cancer Inst. 2002, 94 (6): 454-460.
Slattery ML, Samowitz W, Hoffman M, Ma KN, Levin TR, Neuhausen S: Aspirin, NSAIDs, and colorectal cancer: possible involvement in an insulin-related pathway. Cancer Epidemiol Biomarkers Prev. 2004, 13 (4): 538-545.
Le Marchand L, Donlon T, Hankin JH, Kolonel LN, Wilkens LR, Seifried A: B-vitamin intake, metabolic genes, and colorectal cancer risk (United States). Cancer Causes Control. 2002, 13 (3): 239-248. 10.1023/A:1015057614870.
Sharp L, Little J: Polymorphisms in genes involved in folate metabolism and colorectal neoplasia: a HuGE review. Am J Epidemiol. 2004, 159 (5): 423-443. 10.1093/aje/kwh066.
Chen J, Kyte C, Valcin M, Chan W, Wetmur JG, Selhub J, Hunter DJ, Ma J: Polymorphisms in the one-carbon metabolic pathway, plasma folate levels and colorectal cancer in a prospective study. Int J Cancer. 2004, 110 (4): 617-620. 10.1002/ijc.20148.
Hansen R, Saebo M, Skjelbred CF, Nexo BA, Hagen PC, Bock G, Bowitz Lothe IM, Johnson E, Aase S, Hansteen IL, Vogel U, Kure EH: GPX Pro198Leu and OGG1 Ser326Cys polymorphisms and risk of development of colorectal adenomas and colorectal cancer. Cancer Lett. 2005, 229 (1): 85-91. 10.1016/j.canlet.2005.04.019.
Küry S, Buecher B, Robiou-du-Pont S, Scoul C, Colman H, Lelièvre B, Olschwang S, Le Houérou C, Le Neel T, Faroux R, Ollivry J, Lafraise B, Chupin LD, Bézieau S: The thorough screening of the MUTYH gene in a large French cohort of sporadic colorectal cancers. Genet Test. 2007, 11 (4): 373-9. 10.1089/gte.2007.0029.
Cox DG, Pontes C, Guino E, Navarro M, Osorio A, Canzian F, Moreno V: Polymorphisms in prostaglandin synthase 2/cyclooxygenase 2 (PTGS2/COX2) and risk of colorectal cancer. Br J Cancer. 2004, 91 (2): 339-343.
Halushka MK, Walker LP, Halushka PV: Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin Pharmacol Ther. 2003, 73 (1): 122-130. 10.1067/mcp.2003.1.
Jiang J, Gajalakshmi V, Wang J, Kuriki K, Suzuki S, Nakamura S, Akasaka S, Ishikawa H, Tokudome S: Influence of the C161T but not Pro12Ala polymorphism in the peroxisome proliferator-activated receptor-gamma on colorectal cancer in an Indian population. Cancer Sci. 2005, 96 (8): 507-512. 10.1111/j.1349-7006.2005.00072.x.
Ulrich CM, Bigler J, Sparks R, Whitton J, Sibert JG, Goode EL, Yasui Y, Potter JD: Polymorphisms in PTGS1 (= COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2004, 13 (5): 889-893.
den Dunnen JT, Antonarakis SE: Nomenclature for the description of human sequence variations. Hum Genet. 2001, 109 (1): 121-124. 10.1007/s004390100505.
Rothman KJ: No adjustments are needed for multiple comparisons. Epidemiology. 1990, 1 (1): 43-46.
Storey JD, Tibshirani R: Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003, 100 (16): 9440-9445. 10.1073/pnas.1530509100.
Ye Z, Parry JM: A meta-analysis of 20 case-control studies of the glutathione S-transferase M1 (GSTM1) status and colorectal cancer risk. Med Sci Monit. 2003, 9 (10): SR83-91.
Brennan P: Gene-environment interaction and aetiology of cancer: what does it mean and how can we measure it?. Carcinogenesis. 2002, 23 (3): 381-387. 10.1093/carcin/23.3.381.
Sole X, Guino E, Valls J, Iniesta R, Moreno V: SNPStats: a web tool for the analysis of association studies. Bioinformatics. 2006, 22 (15): 1928-1929. 10.1093/bioinformatics/btl268.
Diaz BL, Arm JP: Phospholipase A(2). Prostaglandins Leukot Essent Fatty Acids. 2003, 69 (2–3): 87-97. 10.1016/S0952-3278(03)00069-3.
Steele VE, Hawk ET, Viner JL, Lubet RA: Mechanisms and applications of non-steroidal anti-inflammatory drugs in the chemoprevention of cancer. Mutat Res. 2003, 523-524: 137-144.
Tan W, Wu J, Zhang X, Guo Y, Liu J, Sun T, Zhang B, Zhao D, Yang M, Yu D, Lin D: Associations of functional polymorphisms in cyclooxygenase-2 and platelet 12-lipoxygenase with risk of occurrence and advanced disease status of colorectal cancer. Carcinogenesis. 2007, 28 (6): 1197-1201. 10.1093/carcin/bgl242.
MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM: The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell. 1995, 81 (6): 957-966. 10.1016/0092-8674(95)90015-2.
Nakanishi M, Rosenberg DW: Roles of cPLA2alpha and arachidonic acid in cancer. Biochim Biophys Acta. 2006, 1761 (11): 1335-1343.
Auwerx J: Nuclear receptors. I. PPAR gamma in the gastrointestinal tract: gain or pain?. Am J Physiol Gastrointest Liver Physiol. 2002, 282 (4): G581-585.
Michalik L, Desvergne B, Wahli W: Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004, 4 (1): 61-70. 10.1038/nrc1254.
Siezen CL, van Leeuwen AI, Kram NR, Luken ME, van Kranen HJ, Kampman E: Colorectal adenoma risk is modified by the interplay between polymorphisms in arachidonic acid pathway genes and fish consumption. Carcinogenesis. 2005, 26 (2): 449-457. 10.1093/carcin/bgh336.
Gong Z, Xie D, Deng Z, Bostick RM, Muga SJ, Hurley TG, Hebert JR: The PPAR{gamma} Pro12Ala polymorphism and risk for incident sporadic colorectal adenomas. Carcinogenesis. 2005, 26 (3): 579-585. 10.1093/carcin/bgh343.
Theodoropoulos G, Papaconstantinou I, Felekouras E, Nikiteas N, Karakitsos P, Panoussopoulos D, Lazaris A, Patsouris E, Bramis J, Gazouli M: Relation between common polymorphisms in genes related to inflammatory response and colorectal cancer. World J Gastroenterol. 2006, 12 (31): 5037-5043.
Gunter MJ, Canzian F, Landi S, Chanock SJ, Sinha R, Rothman N: Inflammation-related gene polymorphisms and colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2006, 15 (6): 1126-1131. 10.1158/1055-9965.EPI-06-0042.
Lee WP, Tai DI, Lan KH, Li AF, Hsu HC, Lin EJ, Lin YP, Sheu ML, Li CP, Chang FY, Chao Y, Yen SH, Lee SD: The -251T allele of the interleukin-8 promoter is associated with increased risk of gastric carcinoma featuring diffuse-type histopathology in Chinese population. Clin Cancer Res. 2005, 11 (18): 6431-6441. 10.1158/1078-0432.CCR-05-0942.
Ulrich CM, Potter JD: Folate and cancer – timing is everything. Jama. 2007, 297 (21): 2408-2409. 10.1001/jama.297.21.2408.
Huang Y, Han S, Li Y, Mao Y, Xie Y: Different roles of MTHFR C677T and A1298C polymorphisms in colorectal adenoma and colorectal cancer: a meta-analysis. J Hum Genet. 2007, 52 (1): 73-85. 10.1007/s10038-006-0082-5.
Hubner RA, Houlston RS: MTHFR C677T and colorectal cancer risk: A meta-analysis of 25 populations. Int J Cancer. 2007, 120 (5): 1027-1035. 10.1002/ijc.22440.
Mu LN, Cao W, Zhang ZF, Cai L, Jiang QW, You NC, Goldstein BY, Wei GR, Chen CW, Lu QY, Zhou XF, Ding BG, Chang J, Yu SZ: Methylenetetrahydrofolate reductase (MTHFR) C677T and A1298C polymorphisms and the risk of primary hepatocellular carcinoma (HCC) in a Chinese population. Cancer Causes Control. 2007, 18 (6): 665-675. 10.1007/s10552-007-9012-x.
Araki A, Hosoi T, Orimo H, Ito H: Association of plasma homocysteine with serum interleukin-6 and C-peptide levels in patients with type 2 diabetes. Metabolism. 2005, 54 (6): 809-814. 10.1016/j.metabol.2005.02.001.
Dedoussis GV, Panagiotakos DB, Pitsavos C, Chrysohoou C, Skoumas J, Choumerianou D, Stefanadis C: An association between the methylenetetrahydrofolate reductase (MTHFR) C677T mutation and inflammation markers related to cardiovascular disease. Int J Cardiol. 2005, 100 (3): 409-414. 10.1016/j.ijcard.2004.08.038.
Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, Brooks LD, Cardon LR, Daly M, Donnelly P, Fraumeni JF, Freimer NB, Gerhard DS, Gunter C, Guttmacher AE, Guyer MS, Harris EL, Hoh J, Hoover R, Kong CA, Merikangas KR, Morton CC, Palmer LJ, Phimister EG, Rice JP, Roberts J, et al: Replicating genotype-phenotype associations. Nature. 2007, 447 (7145): 655-660. 10.1038/447655a.
Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG: Replication validity of genetic association studies. Nat Genet. 2001, 29 (3): 306-309. 10.1038/ng749.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/8/326/prepub
Acknowledgements
We are very grateful to all the patients, control individuals, and physicians who participated in this work and made it possible. This work was funded by a regional Hospital Clinical Research Program (PHRC) and supported by the Regional Council of Pays de la Loire, the Groupement des Entreprises Françaises dans la LUtte contre le Cancer (GEFLUC), the Association Anne de Bretagne Génétique and the Ligue Régionale Contre le Cancer (LRCC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SK, BB, and SRdP equally contributed to this work. SK participated in the design of the study, carried out statistical analyses, contributed to genotyping assays, and drafted the manuscript; BB conceived the study, participated in its design, and coordinated the recruitment of patients and controls; SRdP participated in statistical analyses and genotyping assays, and helped to draft the manuscript; CS did samples management and contributed to genotyping assays; HC contributed to molecular genetic analyses; TLN coordinated data storage and management; CLH coordinated the recruitment of patients and controls; RF and JO contributed to the collection of patients samples; BL and LDC contributed to the collection of controls samples; VS participated in statistical analyses; SB conceived the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Sébastien Küry, Bruno Buecher, Sébastien Robiou-du-Pont contributed equally to this work.
Electronic supplementary material
12885_2008_1264_MOESM3_ESM.xls
Additional file 3: Statistical analysis of association between CRC risk and polymorphisms selected for the study using conditional logistic regression (number of age- and sex-matched pairs = 811, i.e., 1622 individuals). (XLS 48 KB)
12885_2008_1264_MOESM4_ESM.xls
Additional file 4: Test of the different possible models of genotypic combinations related to a given set of five polymorphisms picked among the 52 polymorphisms analysed in the study. (XLS 62 KB)
12885_2008_1264_MOESM5_ESM.xls
Additional file 5: Estimation by use of bootstrapping of the external validity of the model showing association between CRC risk and genotypes combinations. (XLS 334 KB)
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Küry, S., Buecher, B., Robiou-du-Pont, S. et al. Low-penetrance alleles predisposing to sporadic colorectal cancers: a French case-controlled genetic association study. BMC Cancer 8, 326 (2008). https://doi.org/10.1186/1471-2407-8-326
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-8-326