
Abstract
Background
Epigenetic alterations are a hallmark of human cancer. In this study, we aimed to investigate whether aberrant DNA methylation of cancer-associated genes is related to urinary bladder cancer recurrence.
Methods
A set of 4 genes, including CDH1 (E-cadherin), SFN (stratifin), RARB (retinoic acid receptor, beta) and RASSF1A (Ras association (RalGDS/AF-6) domain family 1), had their methylation patterns evaluated by MSP (Methylation-Specific Polymerase Chain Reaction) analysis in 49 fresh urinary bladder carcinoma tissues (including 14 cases paired with adjacent normal bladder epithelium, 3 squamous cell carcinomas and 2 adenocarcinomas) and 24 cell sediment samples from bladder washings of patients classified as cancer-free by cytological analysis (control group). A third set of samples included 39 archived tumor fragments and 23 matched washouts from 20 urinary bladder cancer patients in post-surgical monitoring. After genomic DNA isolation and sodium bisulfite modification, methylation patterns were determined and correlated with standard clinic-histopathological parameters.
Results
CDH1 and SFN genes were methylated at high frequencies in bladder cancer as well as in paired normal adjacent tissue and exfoliated cells from cancer-free patients. Although no statistically significant differences were found between RARB and RASSF1A methylation and the clinical and histopathological parameters in bladder cancer, a sensitivity of 95% and a specificity of 71% were observed for RARB methylation (Fisher's Exact test (p < 0.0001; OR = 48.89) and, 58% and 17% (p < 0.05; OR = 0.29) for RASSF1A gene, respectively, in relation to the control group.
Conclusion
Indistinct DNA hypermethylation of CDH1 and SFN genes between tumoral and normal urinary bladder samples suggests that these epigenetic features are not suitable biomarkers for urinary bladder cancer. However, RARB and RASSF1A gene methylation appears to be an initial event in urinary bladder carcinogenesis and should be considered as defining a panel of differentially methylated genes in this neoplasia in order to maximize the diagnostic coverage of epigenetic markers, especially in studies aiming at early recurrence detection.
Similar content being viewed by others


Background
Urinary bladder cancer is the fourth most common malignancy in the Western world, with a male:female ratio of nearly four to one and a median age at diagnosis between 65 and 70 years [1]. Histologically, 90% to 95% of malignant bladder tumors are urothelial carcinoma (UC), formerly designated transitional cell carcinoma (TCC) [2]. Although more than 70% of the lesions are detected as non-invasive papillary carcinomas, which commonly recur, a poor prognosis is related to tumors that are already invasive at diagnosis (~20%) [3]. After transurethral resection of superficial bladder cancer, periodic cystoscopic monitoring is performed for early recurrence detection, with some cases requiring intravesical prophylactic instillation chemotherapy. Muscle invasive disease calls for more aggressive treatment, often consisting of radical cystectomy and bladder substitution [4].
At present, conventional diagnosis for urinary bladder cancer is based on morphological, histological and pathological features. These criteria provide essential prognostic information, but show insufficient power to precisely predict patient outcome. The need for accurate predictive markers has led to the search for molecular markers in bladder cancer patients [5]. The use of genetic and epigenetic alterations for the early detection of bladder cancer is promising because it is believed that some molecular events occur at the beginning of the carcinogenesis process. Thus, molecular diagnosis may allow detection before clinical or radiographic manifestations. In this context, a sensitive and specific noninvasive test could prescreen patients with clinical symptoms as well as those at high risk, and would also be useful in monitoring patients post-surgically for early detection of recurrence.
DNA-, RNA-based or/and immunohistochemical methods have been applied to identify new tumor markers or to estimate risk of tumor progression in UC. Several DNA alterations have been described in bladder cancer, such as allele losses or deletions [6], gene amplifications [7], DNA mutations [8] and microsatellite instabilities [9]. Furthermore, aberrant DNA methylation patterns have been recognized as common epigenetic changes in human cancer and are already detected in early cancer stages [10]. DNA methylation occurs on cytosine residues located at the 5' position of guanines in CpG dinucleotides [11]. Its distribution on the mammalian genome is not random and is especially important in CpG-rich areas, also called CpG islands. The promoter region of actively transcribing genes is frequently rich in this dinucleotide sequence, almost always unmethylated [12].
Dense DNA methylation in CpG islands of growth-regulating gene promoter regions is now recognized as a common alternative mechanism for gene inactivation in human cancer, an event as important as somatic mutations in coding regions of tumor suppressor genes (TSG) [13]. Usually both genetic and epigenetic events represent complementary hits involved in TSG inactivation [14]. A large number of studies have shown that loci of epigenetically inactivated TSG generally coincide with overlapping regions of allelic losses in human cancer, including UC [6, 15–19]. In fact, loss of heterozygosity (LOH) assays have been widely used as indirect approaches in the search for a new TSG [20, 21]. In the last few years, genetic studies have indicated that allelic loss in many distinct chromosomal regions, including 1p, 3p, and 16q, are associated with UC tumorigenesis [17–19, 21]. It is important to notice that RASSF1A (Ras association (RalGDS/AF-6) domain family 1) and RARB (retinoic acid receptor, beta) mapped at 3p (3p21.3 and 3p24, respectively), SFN (stratifin, also known as 14-3-3σ) located at 1p35.3, and CDH1 (cadherin 1, type 1, E-cadherin [epithelial]) at 16q22.1, are epigenetically silenced TSGs located at loci that overlap with LOH minimal regions in human cancer.
RASSF1A protein probably modulates some of the growth inhibitory responses mediated by Ras, although its interaction with activated Ras remains unclear. This gene is considered a bona fide tumor suppressor epigenetically inactivated during human carcinogenesis, whose hypermethylation has also been reported in UC [22–30]. The RARB gene is a member of the thyroid-steroid hormone receptor superfamily of nuclear transcriptional regulators that binds retinoic acid (the biologically active form of vitamin A), and also mediates cellular signaling during embryonic morphogenesis, cell growth, and differentiation [31]. Retinoic acids exhibit tumor-suppressor activity due to their antiproliferative and apoptosis-inducing effects [32]. RARB has also presented high methylation frequencies in urinary bladder tumors (varying from 15% to 93%) [15, 22, 24, 25, 30].
Initially, it was suggested that loss of stratifin expression could contribute to malignant transformation by disabling the cell cycle arrest at the G2 checkpoint, allowing the accumulation of genetic defects [33]. Subsequently, the down-regulation of SFN gene in various human cancers was generally attributed to the hypermethylation of its CpG island. To the best of our knowledge, there is only one previous study addressing SFN hypermethylation in UC where the highest frequency was found for squamous cell carcinomas irrespective of their grade of cellular malignancy (80%). Furthermore, the authors found hypermethylation of 57.1% for high grade, high stage nonpapillary TCC; 28.6% for low grade, low stage papillary TCC; and 28% for undifferentiated small cell carcinomas, the lowest rate [34].
The CDH1 gene encodes for a calcium-dependent cell-cell adhesion glycoprotein, whose loss of function may contribute to cancer progression by increasing proliferation, invasion, or metastasis [35]. These findings suggest that CDH1 is a tumor- and invasion-suppressor gene [36]. Its relation to urinary bladder carcinogenesis was demonstrated through the observation of altered expression due to epigenetic changes in many studies, ranging from 9.5% hypermethylation [29] up to 87% in TCCs [37]. However, some studies have also evaluated TCCs, both in squamous cells and in situ carcinoma, and have shown a variable spectrum of hypermethylation for the same gene [15, 22, 24, 25, 27, 30, 38–40].
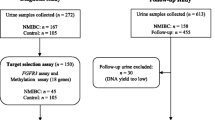
In an effort to identify a possible association among epigenetic changes, urinary bladder cancer prognosis and early-recurrence, we analyzed the methylation pattern of CDH1, RARB, SFN and RASSF1A genes in 54 fresh samples of urinary bladder cancer, 14 of which were paired with tumor-adjacent normal urothelium; 39 paraffin-embedded UC primary tumor and/or recurrence matched with 23 bladder washing sediments obtained from 20 patients under post-surgical monitoring. In addition, we analyzed a hospital-based control group consisting of 24 bladder washings from patients that reported urological complaints, but without any bladder tumor history and showing negative cytology for tumor cell presence.
Methods
Sample collection and DNA extraction
Methylation patterns of RASSF1A, RARB, SFN, and CDH1 genes were determined in two cell lineages, 5637 and T24, derived from non-invasive and invasive high-grade UC, respectively. Fresh samples of tumoral urinary bladder tissues were obtained from 54 patients (44 males and 10 females; median age of 67.85 years, ranging from 40 to 90 years) who underwent surgical treatment at Amaral Carvalho Hospital, Jaú, SP, Brazil. Patients were recruited consecutively on the basis of tissue availability. Treatment for each patient consisted of initial endoscopic tumor resection and subsequent radical cystectomy for those with muscle invasive disease. Non-muscle invasive tumors underwent intravesical bacillus Calmette-Guerin (BCG) therapy. Normal adjacent tissue samples were also collected from each case. A tumor fragment and the matched normal adjacent tissue were fixed in formalin and embedded in paraffin. The corresponding hematoxylin-eosin-stained sections were evaluated by the same pathologist (JLVC) to determine tumor type, grade and growth pattern. Samples were trimmed to maximize the quantity of target tissue and only fragments with more than 70% neoplastic cells were used for DNA extraction. After this evaluation, only 14 normal adjacent tissue samples exhibited an epithelial layer. Tumors were staged according to the 1998 WHO-ISUP classification [41].
A control group included 24 urinary bladder washings from patients that reported urological complaints, but without any bladder tumor history and showing negative cytology for tumor cell presence in the same bladder washing cell samples.
In addition, 20 patients in post-surgical monitoring, who underwent cytology analysis to detect tumor recurrence, were recruited at the Department of Urology from Botucatu Medical School, UNESP – Sao Paulo State University, Brazil. From this group was collected a total of 23 urinary bladder washings matched with 39 UC samples obtained from the Department of Pathology archive. Among these 20 patients 9 presented recurrent tumors (analysis until seven biopsies had been collected at distinct times) and 11 were primary tumors.
Genomic DNA from fresh bladder tissues, paraffin-embedded samples and washout cell sediments were obtained by standard sodium dodecyl sulfate/proteinase K digestion, followed by phenol/chloroform extraction and ethanol precipitation.
All samples were collected after patients or their relatives had provided informed consent. Approval for research on human subjects was obtained from the respective ethic committees of both institutions and by the National Research Ethics Committee (CONEP 9382) Brasilia, DF, Brazil.
Bisulfite treatment and Methylation-Specific Polymerase Chain Reaction (MSP)
The conversion of DNA by sodium bisulfite was performed using an established protocol [42] with modifications. Initially, genomic DNA was denatured with 3 M NaOH at 40°C for 15 min (final concentration of 0.3 M NaOH). The urea/bisulfite and hydroquinone solution (freshly prepared, pH 5.0) were then added to the denatured DNA to yield final concentrations of 5.36 M, 3.44 M, and 0.5 mM, respectively, followed by 20 cycles of incubation at 55°C for 15 min followed by denaturation at 95°C for 30 sec in a PTC200 Peltier Termal Cycler (MJ Research, Madison, USA). DNA was purified with the Wizard DNA Clean-UP System (Promega. Madison, WI, USA), and DNA modification was completed by the addition of 5.0 μl of NaOH 3 M at room temperature for 15 min. Precipitation was carried out through the addition of 30 μl of ammonium acetate 5 M (pH 7.0), 350 μl of ethanol and 1 μl of glycogen (20 μg/uL) (Invitrogen Life Technologies, Carlsbad, CA, USA). The bisulfite-modified DNA was resuspended in 20 μl of sterile water and stored at -20°C.
The methylation pattern of promoter regions for CDH1, RARB, SFN and RASSF1A genes was evaluated by a MSP approach. For each gene, previously described primers specific to the methylated and unmethylated sequences were used [33, 43–45]. DNA from lymphocytes of healthy volunteers treated with SssI methyltransferase (New England Biolabs, Beverly, MA, USA) and then subjected to bisulfite modification was used as positive controls for methylated alleles. The reaction was performed in a total volume of 50 μl containing 10 μg of genomic DNA, 10 U of SssI methylase, 160 mM of S-adenosyl-metionina, 50 mM of NaCl, 10 mM of Tris-HCl, 10 mM of MgCl2, 1 mM of DTT pH 7.9, during 18 hours at 37°C.
Table 1 summarizes the oligonucleotide sequences, annealing temperature and product size for MSP analysis. To determine the methylation pattern within the CpG island in 5'UTR of the CDH1 gene, a nested-PCR approach was used as previously described in detail [46].
One-step MSP was performed to detect the methylation pattern of RARB, SFN and RASSF1A genes, using specific primers for the methylated and unmethylated sequences in distinct reactions, accomplished in a total volume of 25 μl containing 0.25 μM of each primer, 200 μM of each dNTP, 15 mM Tris-HCl, pH 8.0, 50 mM KCl, 1U of AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA) and 3 mM MgCl2 for RARB and RASSF1A, and 2.5 mM MgCl2 for SFN.
The amplified products were visualized after electrophoresis in 6% polyacrylamide gel and silver nitrate staining [47]. Water blanks were included in each assay.
Statistical analysis
Descriptive mean and percentage statistics were used to summarize patient data and gene hypermethylation status. The presence of methylation and characteristics including age, sex and clinico-histopathological parameters were evaluated using Odds Ratio (OR) with Confidence Interval (CI) of 95%. Pairwise associations followed dichotomous variables defined according to growth pattern (non-papillary versus papillary), differentiation grade (low versus high), tumor invasiveness (noninvasive versus invasive) [41], and presence or absence of tumoral recurrence. Potential associations on the presence of promoter methylation for each gene as well as the sensitivity and specificity of the assay for tumor recurrence were assessed using Fisher's Exact test with a 5% significance level. Correlations between cytology and hypermethylation of bladder washings were considered to assess the relative hazar of recurrence. All statistical evaluations were performed using a computer-assisted program (SPSS – Statistical Package for The Social Sciences v15.0, SPSS Inc.).
Results
MSP analysis in cell lineages
MSP analysis of DNA from 5637 and T24 cell lines evidenced hypermethylation at the CDH1 and RASSF1A gene promoter regions. None of them showed this pattern for the RARB gene. The methylation of SFN was observed in 5637 cells, but was not present in T24.
MSP analysis in matched tumoral and adjacent bladder tissue samples
Fifty-four matched tumoral and adjacent tissue samples were collected. Remarkably, after histopathology, the presence of normal epithelial cell layer in normal adjacent biopsies was confirmed in only 14 pairs. Thus, MSP analysis was performed on 49 UCs obtained from unrelated patients, 14 of them matched with normal tissue samples, 3 squamous cell carcinomas and 2 adenocarcinomas. Table 2 summarizes the relevant clinical and histopathological characteristics in the group of 49 UC patients. On average, these patients received 30 months of follow-up monitoring. Twenty-five UC patients showed recurrence: 20 (40.8%) were recurrent at the moment of the study sample collection, since they had a positive history of previous UC confirmed by a histological diagnosis before the date of the most recent surgery (0.7 to 7.5 years); and 5 had their primary tumor evaluated, but exhibited recurrence within a short time period (Table 3).
After genomic DNA treatment with sodium bisulfite and MSP analysis, both amplicons for unmethylated and methylated alleles, respectively, were detected in CDH1 and SFN gene target regions in all 49 fresh UC samples. Among histopathologically normal tumor-adjacent urinary bladder tissues, the same methylation pattern was found, except in one sample which showed only the unmethylated alleles for SFN gene.
RARB and RASSF1A hypermethylation were detected in 40 (81.6%) and 11 (22.5%) UC samples, respectively. The comparison of 14 matched normal and tumoral urinary bladder samples exhibited a concordant pattern for presence of RARB hypermethylation in 12 pairs; in two pairs (cases 12 and 42, Figure 1) RARB hypermethylation was restricted to normal tissue. Absence of RASSF1A hypermethylation was a common feature in 9 bladder tissue pairs. Only one case showed hypermethylation in both normal and tumoral tissues (case 11, Figure 1) and, in 3 matched samples (cases 22, 29 and 36, Figure 1) it was restricted to the tumor specimens. Adjusted ORs for these data as well as demographic, physician and histopathological parameters related to the RARB and RASSF1A DNA methylation patterns in 49 UC samples are shown in Table 3.

CDH1, RARB, SFN and RASSF1A methylation patterns observed in fresh UC samples paired with normal adjacent urinary bladder tissue. M – methylated allele; U – unmethylated allele; N – normal tissue; T – tumoral tissue.
The adenocarcinomas analyzed showed hypermethylation for both, RARB and RASSF1A genes. Two of the squamous cell carcinomas showed the same methylation pattern for RARB, and one for RASSF1A.
MSP analysis in exfoliated cells (bladder washings) in cancer-free controls and in bladder cancer patients
In the control group, of the 24 urinary bladder exfoliated cell samples submitted to cytological analysis from patients (median age of 61.4, ranging from 26 to 82 years) with negative diagnosis for cancer (four of them with cystitis), 2/23 (8.3%) and 8/16 (50%) exhibited RARB and RASSF1A hypermethylation, respectively. In addition, higher frequencies of methylation were detected for CDH1 (91.3%) and SFN (95.5%) genes. Compared to the results shown by the biopsy analysis of UCs, RARB had a sensitivity of 95% and specificity of 71% by Fisher's Exact test (p < 0.0001; OR = 48.89); for the same parameters, RASSF1A showed 58% and 17%, respectively (p < 0.05; OR = 0.29) (Table 4).
In the third sample set, hypermethylation identified in tumor DNA from archival UC samples was used as a molecular tag to predict tumor recurrence in the corresponding DNA obtained from cells of urinary bladder washings from UC patients under post-surgical monitoring. The comparative analysis between MSP from washout cells and corresponding primary and/or recurrence tumor sample was done for RARB and RASSF1A genes, including 23 urinary bladder washings and 39 paraffin-embedded UC samples from 20 patients (median age of 68.65, ranging from 42 to 84 years) (Figure 2). Among 39 tissue samples, RARB hypermethylation was identified in 14/38 fragments (36.8%) analyzed. Twelve patients showed at least one tumor fragment hypermethylated for RARB gene. Due to limited tissue volume, RASSF1A gene was analyzed in a subset of 15 fragments from 11 patients, with methylation detected in 9/15 of them (60%); these hypermethylated tumors were from 7 patients.

A) Cytological analysis of bladder washing sediment negative for the presence of tumor cells (case 11). B) Positive cytology illustrating a tumor recurrent case during the post-surgical monitoring. × 400, Giemsa staining. C) Comparative MSP results from case 11 in tumor tissue (TCC) and in the exfoliated cells from the correspondent bladder washing for RARB and RASSF1A genes. M – methylated allele; U – unmethylated allele. D) Distribution of MSP results among the third set of samples including 39 archived tumor fragments and 23 matched washouts from 20 urinary bladder cancer patients in post-surgical monitoring and comparison with the cytological analysis.
Patients showing at least one hypermethylated tumor fragment for RARB and/or RASSF1A were considered informative cases for further comparisons. The MSP results were challenged by cytological analysis during the post-surgical monitoring: 5/12 patients showed positive cytology at the time of cell collection to MSP analysis, but only one of these exhibited RARB hypermethylation in the same washout cells. MSP analysis results of RASSF1A gene in 7 informative patients were discordant because none of the 4 recurrent cases by cytological analysis showed the epigenetic marker; contrarily, RASSF1A hypermethylation was detected in 2 of the 3 non-recurrent cases. Table 5 exhibits the sensitivity and specificity of MSP analysis in relation to the gold-standard cytological evaluation for tumor recurrence detection.
Discussion
Epigenetic alterations are a hallmark of human cancer. In particular, DNA hypermethylation is a common mechanism for inactivating tumor-suppressor and other cancer genes in tumor cells [48]. The aberrant methylation patterns have been used as targets for the detection of tumor cells in clinical specimens such as tissue biopsies or body fluids [49].
In our MSP analysis performed on DNA obtained from fresh tumor samples, a hypermethylated pattern predominated for CDH1 and SFN genes. Commonly, a large spectrum of hypermethylation frequencies has been reported for several genes in bladder cancer. For example, CDH1 gene methylation frequencies range from 9.5% to 84% [15, 22, 25, 27, 30, 37–40], independently of histological classification. We detected CDH1 hypermethylation frequency of 100% in bladder UCs, squamous cell carcinomas and adenocarcinomas samples, as well as in normal adjacent urinary bladder tissue and in exfoliated urothelial cells from cancer-free controls. Similarly, the SFN gene was also hypermethylated in these samples. Costa et al. [50] also detected high frequencies of CDH1 methylation in clear cell renal carcinomas and normal renal tissues (82.7% and 87.1%, respectively); in addition, SFN was hypermethylated in 100% of normal and tumoral renal tissues analyzed.
Interpretation of differential DNA-methylation patterns in cancer has proven difficult, in part because the functional consequences depend on the genomic region involved, the specific CpG dinucleotides, and the inter- and intratumoral heterogeneity. Apart from this, methodological issues such as the different primer sets interrogating methylation at distinct CpG dinucleotides of a specific promoter region could explain the range of frequencies reported in the literature. In our study, the protocol used (which included the addition of urea in the DNA modification step in order to improve the efficiency of unmethylated cytosine conversion [42]) and the CDH1 gene analysis based on the nested-PCR approach may have contributed to the high methylation prevalence observed.
The dynamic nature of epigenetic alterations is partially due to polymorphisms in some methyl group metabolism genes [51, 52] and in genes coding for proteins that mediate these changes (DNA methyltransferases, methyl-CpG-binding domain proteins) [53]. In addition, genomic profiles of DNA methylation are also influenced by aging [38, 54], dietary intake [55, 56] and environmental exposure [53, 57, 58]. In this context, DNA methylation heterogeneous patterns should be expected and already detected for some genomic regions as reported by Eckkhardt et al. [59]. These authors have found that 30.2% of investigated loci on chromosomes 6, 20, and 22 exhibit heterogeneity of methylation status, mainly due to the mosaic patterns in the studied tissue. Furthermore, 10% of the analyzed regions showed tissue-specific differences in DNA methylation.
We observed hypermethylated CDH1, SFN, and RARB genes in the normal-adjacent tissue of urinary bladder tumor. Aberrant methylation patterns have been associated with chronic inflammation [60, 61], viral infection [62] and aging [63]. Smith and Pereira-Smith [64] have previously reported that epigenetic alterations are involved in both the etiology and consequences of aging. Thus, hypermethylation in normal tissue as detected in the present study agrees with the results previously found by Bornman et al. [38], who observed a similar pattern for CDH1 in normal bladder tissue from patients older than 70 years. Furthermore, aberrant methylation patterns of the CDH1 promoter region were also described in pre-adenoma stages of colorectal cancer, in hyperplasic polyps [65, 66] and in ulcerative colitis (a chronic inflammatory condition of the large intestine that predisposes to cancer) [60, 67]. During breast cancer progression, CDH1 gene methylation occurs in about 30% of the ductal carcinomas in situ, with a significant increase in invasive and metastatic lesions [68]. Moreover, this gene has been also found methylated in pre-malignant and invasive bladder cancers. In mammary tissue, SFN is usually unmethylated in normal epithelium, but methylated in atypical hyperplasias, intraductal papillomas, ductal in situ carcinomas, infiltrating carcinomas and in stromal cells [69, 70]. SFN and CDH1 methylation have been reported in peripheral blood cells [70] as well as in infiltrating leukocytes in breast cancer [71]. Overall, these observations suggest that both genes, CDH1 and SFN, are not effective biomarkers for MSP analysis in bladder cancer.
The MSP analysis of RARB and RASSF1A genes showed respective hypermethylation of 82.9% and 24.4% in 49 UCs analyzed. Investigating the methylation at the same CpG dinucleotides, Maruyama et al. [22], and Catto et al. [25], found 15% and 24% hypermethylation for RARB, and 35% and 54% for RASSF1A, respectively. In order to verify the specificity of RARB and RASSF1A hypermethylation in relation to malignant phenotype in bladder tissue, we evaluated these genes in exfoliated urothelial cells from patients without cancer (control group): RARB and RASSF1A hypermethylation were detected in 8.3% and 33.3%, respectively. The comparison of these data revealed that RARB hypermethylation provides better diagnostic coverage and specificity than RASSF1A hypermethylation. However, the hypermethylated pattern of these genes in normal adjacent tissue in matched samples, especially for the RARB gene, was an unexpected finding. In this context, we could hypothesize that molecular alterations precede morphological changes in the exposed urinary bladder epithelium, since patients with bladder neoplasia frequently show genetic instability on apparently normal mucosa besides alterations of surrounding tissue [72]. Aberrant methylation patterns appear to reflect a pre-malignant characteristic of the urinary bladder mainly because UC is a neoplasia with multifocal lesions and elevated recurrence indices [73], thus corroborating the hypothesis that epigenetic alterations in cancer may preexist in morphologically normal cells [74].
Thus, genetic and epigenetic alterations may be present before cancer detection by imaging or traditional pathology investigations. Therefore, molecular tests that target these alterations have conceptual advantages for the successful early detection of neoplasias [48]. DNA represents an ideal substrate for molecular detection because it is robust, survives adverse conditions that many clinical specimens undergo and, can be readily amplified by powerful PCR-based approaches [75]. Tiny amounts of DNA from early pre-neoplastic lesions or small cancers can be used to permit the sensitive detection of one cancer cell in a background of hundreds of normal cells.
Hence, the predictive value of MSP in identifying tumor cells in washing sediments was evaluated in bladder cancer patients under post-surgical monitoring to detect tumor recurrence. Positive cytology was found in 33.3% of patients with urinary bladder tumor history. The hypermethylation patterns of RARB and RASSF1A genes observed in cells obtained from urinary bladder washing sediments were not concordant: some hypermethylated cases in tumor tissue and recurrence by cytological analysis did not show this marker in the same exfoliated cells. Contrarily, in four cases, the cells taken from urinary bladder washings exhibiting hypermethylation for these genes did not match the hypermethylation of the correspondent TCC. The heterogeneity of the intra- and intertumoral methylation patterns could partially explain these discrepancies. Thus, hypermethylation could already be present in the urinary bladder epithelium of cancer patients but not necessarily in cells exfoliated from the urinary bladder of cancer-free patients. RARB hypermethylation confirmed the presence of tumor cells in only 1 out of 5 recurrent cases and was absent in all cases showing negative cytology. Importantly, for the eight patients whose tumors did not present RARB methylation, the paired cell washing sediments DNA were also negative for methylation. This finding corroborates the idea that the methylation pattern of this gene is specific for tumor cells. RASSF1A gene MSP analysis in washout cells showed discordant results since its hypermethylation was not detected in 4 recurrent cases, although 2 negative cases for tumor cells using cytology showed this tumor tag, which suggest that these patients are under high risk for tumor recurrence. Some studies using promoter hypermethylation identified in tumor DNA as a target for cancer detection in the correspondent urine sample have shown sensitivities ranging from 49% to 91% [15, 26, 30, 75]. Recently, Yu et al. [76] included the RASSF1A in an 11-gene set to assessment of DNA methylation in urine sediments for sensitive/specific detection of bladder cancer. Although two studies have reported that the overall methylation sensitivity was significantly higher than cytology [15, 77], several factors may contribute to the lower sensitivities of MSP analysis in cells from urinary bladder fluids including the incomplete diagnostic coverage of selected gene sets, limited quantity of cells sampled, and the intrinsic heterogeneity of methylation patterns in the exposed epithelium, among others.
Conclusion
In the literature, no single gene was found to be consistently methylated in most bladder tumors. Thus, panels of genes that are methylated in urinary bladder cancer have been investigated to define methylation profiles associated with urinary bladder cancer diagnosis, prognosis and early recurrence detection. DNA hypermethylation of CDH1 and SFN genes was detected indistinctly among urinary bladder tumoral and normal tissues as well as in urinary bladder exfoliated cells, suggesting that these epigenetic features do not satisfy enough specificity criteria for use as prognostic or early detection markers. The methylation of RARB and RASSF1A genes appears to be an initial event in urinary bladder carcinogenesis maintained during tumor progression and should be included in the panels of differentially methylated genes in urinary bladder cancer in order to maximize the diagnostic coverage of epigenetic markers.
Abbreviations
- CDH1 :
-
cadherin 1, type 1, E-cadherin [epithelial] gene
- LOH :
-
loss of heterozygosity
- MSP :
-
methylation-specific PCR
- RARB :
-
retinoic acid receptor, beta gene
- RASSF1A :
-
Ras association (RalGDS/AF-6) domain family 1 gene
- SFN :
-
stratifin, also know as 14-3-3σ gene
- TCC :
-
transitional cell carcinoma
- TSG:
-
tumor suppressor gene
- UC :
-
urothelial carcinoma.
References
Kirkali Z, Chan T, Manoharan M, Algaba F, Bush C, Cheng L, Kiemeney L, Kriegmair M, Montinroni R, Murphy WM, Sesterhenn IA, Tachibana M, Weider J: Bladder Cancer: Epidemiology, staging and grading, and diagnosis. Urology. 2005, 66: 4-34. 10.1016/j.urology.2005.07.062.
Reuter VE: The pathology of bladder cancer. Urology. 2006, 67: 11-17. 10.1016/j.urology.2006.01.037.
Knowles MA: Tumor suppressor loci in bladder cancer. Front Biosci. 2007, 12: 2233-2251. 10.2741/2226.
Christoph F, Weikert S, Kempkensteffen C, Krause H, Schostak M, Miller K, Schrader M: Regularly methylated novel pro-apoptotic genes associated with recurrence in transitional cell carcinoma of the bladder. Int J Cancer. 2006, 119: 1396-1402. 10.1002/ijc.21971.
Blaveri E, Simko JP, Korkola JE, Brewer JL, Baehner F, Mehta K, Devries S, Koppie T, Pejavar S, Carroll P, Waldman FM: Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res. 2005, 11: 4044-4055. 10.1158/1078-0432.CCR-04-2409.
Bartoletti R, Cai T, Nesi G, Roberta Girardi L, Baronti G, Dal Canto M: Loss of P16 Expression and Chromosome 9p21 LOH in Predicting Outcome of Patients Affected by Superficial Bladder Cancer. J Surg Res. 2007, 143: 422-427. 10.1016/j.jss.2007.01.012.
Leonardo C, Merola R, Orlandi G, Leonardo F, Rondoni M, De Nunzio C: C-erb-2 gene amplification and chromosomal anomalies in bladder cancer: preliminary results. J Exp Clin Cancer Res. 2005, 24: 633-638.
Cho HY, Park HS, Lin Z, Kim I, Joo KJ, Cheon J: BCL6 gene mutations in transitional cell carcinomas. J Int Med Res. 2007, 35: 224-230.
Turyn J, Matuszewski M, Schlichtholz B: Genomic instability analysis of urine sediment versus tumor tissue in transitional cell carcinoma of the urinary bladder. Oncol Rep. 2006, 15: 259-265.
Baylin SB, Ohm JE: Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?. Nat Rev Cancer. 2006, 6: 107-116. 10.1038/nrc1799.
Herman JG, Graff JR, Myohanen S, Nelkinm BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996, 93: 9821-9826. 10.1073/pnas.93.18.9821.
Turker MS, Bestor TH: Formation of methylation patterns in the mammalian genome. Mutat Res. 1997, 386: 119-130. 10.1016/S1383-5742(96)00048-8.
Jones PA, Baylin SB: The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002, 3: 415-428. 10.1038/nrg962.
Lodygin D, Epanchintsev A, Menssen A, Diebold J, Hermeking H: Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 2005, 65: 4218-4227. 10.1158/0008-5472.CAN-04-4407.
Chan MW, Chan LW, Tang NL, Tong JH, Lo KW, Lee TL, Cheung HY, Wong WS, Chan PS, Lai FM, To KF: Hypermethylation of multiple genes in tumor tissues and voided urine in urinary bladder cancer patients. Clin Cancer Res. 2002, 8: 464-470.
Esteller M: CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002, 21: 5427-5440. 10.1038/sj.onc.1205600.
Horikawa Y, Sugano K, Shigyo M, Yamamoto H, Nakazono M, Fujimoto H, Kanai Y, Hirohashi S, Kakizoe T, Habuchi T, Kato T: Hypermethylation of an E-cadherin (CDH1) promoter region in high grade transitional cell carcinoma of the bladder comprising carcinoma in situ. J Urol. 2003, 169: 1541-1545. 10.1097/01.ju.0000046242.55722.1c.
Tzai TS, Chen HH, Chan SH, Ho CL, Tsai YS, Cheng HL, Dai YC, Lin JS, Yang WH, Chow NH: Clinical significance of allelotype profiling for urothelial carcinoma. Urology. 2003, 62: 378-384. 10.1016/S0090-4295(03)00344-3.
Zhang J, Zheng S, Gao Y, Rotolo JA, Xiao Z, Li C, Cheng S: A partial allelotyping of urothelial carcinoma of bladder in the Chinese. Carcinogenesis. 2004, 25: 343-347. 10.1093/carcin/bgh015.
Weinberg RA: Tumor supressor genes. Science. 1991, 254: 1138-1146. 10.1126/science.1659741.
Hoque MO, Lee CC, Cairns P, Schoenberg M, Sidransky D: Genome-wide genetic characterization of bladder cancer: a comparison of high-density single-nucleotide polymorphism arrays and PCR-based microsatellite analysis. Cancer Res. 2003, 63: 2216-2222.
Maruyama R, Toyooka S, Toyooka KO, Harada K, Virmani AK, Zöchbauer-Müller S, Farinas AJ, Vakar-Lopez F, Minna JD, Sagalowsky A, Czerniak B, Gazdar AF: Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001, 61: 8659-8663.
Chan MW, Chan LW, Tang NL, Lo KW, Tong JH, Chan AW, Cheung HY, Wong WS, Chan PS, Lai FM, To KF: Frequent hypermethylation of promoter region of RASSF1AA in tumor tissues and voided urine of urinary bladder cancer patients. Int J Cancer. 2003, 104: 611-616. 10.1002/ijc.10971.
Gutierrez MI, Siraj AK, Khaled H, Koon N, El-Rifai W, Bhatia K: CpG island methylation in Schistosoma- and non-Schistosoma-associated bladder cancer. Mod Pathol. 2004, 17: 1268-1274. 10.1038/modpathol.3800177.
Catto JW, Azzouzi AR, Rehman I, Feeley KM, Cross SS, Amira N, Fromont G, Sibony M, Cussenot O, Meuth M, Hamdy FC: Promoter hypermethylation is associated with tumor location, stage, and subsequent progression in transitional cell carcinoma. Clin Oncol. 2005, 23: 2903-2910. 10.1200/JCO.2005.03.163.
Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, Toma MI, Huland H, Yoo C, Tsai YC, Nichols PW, Bochner BH, Jones PA, Liang G: Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin Cancer Res. 2004, 10: 7457-7465. 10.1158/1078-0432.CCR-04-0930.
Friedrich MG, Chandrasoma S, Siegmund KD, Weisenberger DJ, Cheng JC, Toma MI, Huland H, Jones PA, Liang G: Prognostic relevance of methylation markers in patients with non-muscle invasive bladder carcinoma. Eur J Cancer. 2005, 41: 2769-2778. 10.1016/j.ejca.2005.07.019.
Dammann R, Schagdarsurengin U, Seidel C, Strunnikova M, Rastetter M, Baier K, Pfeifer GP: The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol. 2005, 20: 645-663.
Marsit CJ, Karagas MR, Danaee H, Liu M, Andrew A, Schned A, Nelson HH, Kelsey KT: Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. 2006, 27: 112-116. 10.1093/carcin/bgi172.
Hoque MO, Begum S, Topaloglu O, Chatterjee A, Rosenbaum E, Van Criekinge W, Westra WH, Schoenberg M, Zahurak M, Goodman SN, Sidransky D: Quantitation of promoter methylation of multiple genes in urine DNA and bladder cancer detection. J Natl Cancer Inst. 2006, 98: 996-1004.
Soprano DR, Qin P, Soprano KJ: Retinoic acid receptors and cancers. Annu Rev Nutr. 2004, 24: 201-221. 10.1146/annurev.nutr.24.012003.132407.
Brtko J: Role of retinoids and their cognate nuclear receptors in breast cancer chemoprevention. Cent Eur J Public Health. 2007, 15: 3-6.
Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, Marks JR, Lambers AR, Futreal A, Stampfer MR, Sukumar S: High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci USA. 2000, 97: 6049-6054. 10.1073/pnas.100566997.
Kunze E, Wendt M, Schlott T: Promoter hypermethylation of the 14-3-3 sigma, SYK and CAGE-1 genes is related to the various phenotypes of urinary bladder carcinomas and associated with progression of transitional cell carcinomas. Int J Mol Med. 2006, 18: 547-557.
Hirohashi S, Kanai Y: Cell adhesion system and human cancer morphogenesis. Cancer Sci. 2003, 94: 575-581. 10.1111/j.1349-7006.2003.tb01485.x.
Qian ZR, Sano T, Yoshimoto K, Asa SL, Yamada S, Mizusawa N, Kudo E: Tumor-specific downregulation and methylation of the CDH13 (H-cadherin) and CDH1 (E-cadherin) genes correlate with aggressiveness of human pituitary adenomas. Mod Pathol. 2007, 20: 1269-1277. 10.1038/modpathol.3800965.
Dhawan D, Hamdy FC, Rehman I, Patterson J, Cross SS, Feeley KM, Stephenson Y, Meuth M, Catto JW: Evidence for the early onset of aberrant promoter methylation in urothelial carcinoma. J Pathol. 2006, 209: 336-343. 10.1002/path.1991.
Bornman DM, Mathew S, Alsruhe J, Herman JG, Gabrielson E: Methylation of the E-cadherin gene in bladder neoplasia and in normal urothelial epithelium from elderly individuals. Am J Pathol. 2001, 159 (3): 831-835.
Ribeiro-Filho LA, Franks J, Sasaki M, Shiina H, Li L-C, Nojima D, Arap S, Carroll P, Enokida H, Nakagawa M, Yonezawa S, Dahiya R: CpG hypermethylation of promoter region and inactivation of E-cadherin gene in human bladder cancer. Mol Carcinog. 2002, 34: 187-198. 10.1002/mc.10064.
Horikawa Y, Sugano K, Shigyo M, Yamamoto H, Nakazono M, Fujimoto H, Kanai Y, Hirohashi S, Kakizoe T, Habuchi T, Kato T: Hypermethylation of an E-cadherin (CDH1) promoter region in high grade transitional cell carcinoma of the bladder comprising carcinoma in situ. J Urol. 2003, 169: 1541-1545. 10.1097/01.ju.0000046242.55722.1c.
Epstein JI, Amin MB, Reuter VR, Mostofi FK: The World Health Organization/International Society of Urological Pathology Consensus Classification of Urothelial (Transitional Cell) Neoplasms of the Urinary Bladder. Bladder Consensus Conference Committee. Am J Surg Path. 1998, 22: 1435-1448. 10.1097/00000478-199812000-00001.
Paulin R, Grigg GW, Davey MW, Piper AA: Urea improves efficiency of bisulphite-mediated sequencing of 5'-methylcytosine in genomic DNA. Nucleic Acids Res. 1998, 26: 5009-5010. 10.1093/nar/26.21.5009.
Corn PG, Heath EI, Heitmiller R, Fogt F, Forastiere AA, Herman JG, Wu TT: Frequent hypermethylation of the 5'CpG island of E-cadherin in esophageal adenocarcinoma. Clin Cancer Res. 2001, 7: 2765-2769.
Evron E, Dooley WC, Umbricht CB, Rosenthal D, Sacchi N, Gabrielson E, Soito AB, Hung DT, Ljung B-M, Davidson NE, Sukumar S: Detection of breast cancer cells in ductal lavage fluid by methylation-specific PCR. Lancet. 2001, 357: 1335-1336. 10.1016/S0140-6736(00)04501-3.
Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD: Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001, 93: 691-699. 10.1093/jnci/93.9.691.
Caldeira JR, Prando EC, Quevedo FC, Neto FA, Rainho CA, Rogatto SR: CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer. 2006, 6: 48-10.1186/1471-2407-6-48.
Sanguinetti CJ, Dias Neto E, Simpson AJG: Rapid silver staining and recovery of PCR products separated on polyacrylamide gels. BioTech. 1994, 17: 914-921.
Cairns P: Gene methylation and early detection of genitourinary cancer: the road ahead. Nat Rev Cancer. 2007, 7: 531-543. 10.1038/nrc2170.
Sidransky D: Emerging molecular markers of cancer. Nat Rev Cancer. 2002, 2: 210-219. 10.1038/nrc755.
Costa VL, Henrique R, Ribeiro FR, Pinto M, Oliveira J, Lobo F, Teixeira MR, Jerónimo C: Quantitative promoter methylation analysis of multiple cancer-related genes in renal cell tumors. BMC Cancer. 2007, 7: 133-10.1186/1471-2407-7-133.
Chan EC, Lam SY, Fu KH, Kwong YL: Polymorphisms of the GSTM1, GSTP1, MPO, XRCC1, and NQO1 genes in Chinese patients with non-small cell lung cancers: relationship with aberrant promoter methylation of the CDKN2A and RARB genes. Cancer Genet Cytogenet. 2005, 162: 10-20. 10.1016/j.cancergencyto.2005.03.008.
Kang S, Kim JW, Kang GH, Park NH, Song YS, Kang SB, Lee HP: Polymorphism in folate- and methionine-metabolizing enzyme and aberrant CpG island hypermethylation in uterine cervical cancer. Gynecol Oncol. 2005, 96: 173-180. 10.1016/j.ygyno.2004.09.031.
Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C: Cancer genetics of epigenetic genes. Hum Mol Genet. 2007, 16 (Spec No 1): R28-49. 10.1093/hmg/ddm021.
Kwabi-Addo B, Chung W, Shen L, Ittmann M, Wheeler T, Jelinek J, Issa JP: Age-related DNA methylation changes in normal human prostate tissues. Clin Cancer Res. 2007, 13: 3796-3802. 10.1158/1078-0432.CCR-07-0085.
Feil R: Environmental and nutritional effects on the epigenetic regulation of genes. Mutat Res. 2006, 600: 46-57.
Waterland RA: Assessing the effects of high methionine intake on DNA methylation. J Nutr. 2006, 136: 1706S-1710S.
Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS: Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007, 67: 876-880. 10.1158/0008-5472.CAN-06-2995.
Sathyanarayana UG, Moore AY, Li L, Padar A, Majmudar K, Stastny V, Makarla P, Suzuki M, Minna JD, Feng Z, Gazdar AF: Sun exposure related methylation in malignant and non-malignant skin lesions. Cancer Lett. 2007, 245: 112-120. 10.1016/j.canlet.2005.12.042.
Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S: DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006, 38: 1378-1385. 10.1038/ng1909.
Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R: Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998, 58: 3942-3945.
Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA: Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001, 61: 3573-3577.
Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY: Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. 2002, 160: 787-794.
Waki T, Tamura G, Sato M, Motoyama T: Age-related methylation of tumor suppressor and tumor-related genes: an analysis of autopsy samples. Oncogene. 2003, 22: 4128-4133. 10.1038/sj.onc.1206651.
Smith JR, Pereira-Smith OM: Replicative senescence: implications for in vivo aging and tumor suppression. Science. 1996, 273: 63-67. 10.1126/science.273.5271.63.
Chan AO, Broaddus RR, Houlihan PS, Issa Jp, Hamilton SR, Rashid A: CpG island methylation in aberrant crypt foci of the colorectum. Am J Pathol. 2002, 160: 1823-1830.
Jass JR, Young J, Leggett BA: Evolution of colorectal cancer: change of pace and change of direction. J Gastroenterol Hepatol. 2002, 17: 17-26. 10.1046/j.1440-1746.2002.02635.x.
Azarschab P, Porschen R, Gregor M, Blin N, Holzmann K: Epigenetic control of the E-cadherin gene (CDH1) by CpG methylation in colectomy samples of patients with ulcerative colitis. Genes Chromosomes Cancer. 2002, 35: 121-126. 10.1002/gcc.10101.
Nass SJ, Herman JG, Gabrielson E, Iversen PW, Parl FF, Davidson NE, Graff JR: Aberrant methylation of the estrogen receptor and E-cadherin 50 CpG islands increases with malignant progression in human breast cancer. Cancer Res. 2000, 60: 4346-4348.
Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J, Sukumar S: Hypermethylation of 14-3-3σ (stratifin) is an early event in breast cancer. Oncogene. 2001, 20: 3348-3353. 10.1038/sj.onc.1204438.
Bhatia K, Siraj AK, Hussain A, Bu R, Gutierrez MI: The tumor suppressor gene 14-3-3σ is commonly methylated in normal and malignant lymphoid cells. Cancer Epidemiol Biomarkers Prev. 2003, 12: 165-169.
Lombaerts M, Middeldorp JW, Weide van der E, Philippo K, van Wezel T, Smit VT, Cornelisse CJ, Cleton-Jansen AM: Infiltrating leukocytes confound the detection of E-cadherin promoter methylation in tumors. Biochem Biophys Res Commun. 2004, 319: 697-704. 10.1016/j.bbrc.2004.05.041.
Cianciulli AM, Leonardo C, Guadagni F, Marzano R, Iori F, De Nunzio C, Franco G, Merola R, Laurenti C: Genetic instability in superficial bladder cancer and adjacent mucosa: an interphase cytogenetic study. Hum Pathol. 2003, 34: 214-221. 10.1053/hupa.2003.30.
Trkova M, Babjuk M, Duskova J, Benesova-Minarikova L, Soukup V, Mares J, Minarik M, Sedlacek Z: Analysis of genetic events in 17p13 and 9p21 regions supports predominant monoclonal origin of multifocal and recurrent bladder cancer. Cancer Lett. 2006, 242: 68-76. 10.1016/j.canlet.2005.10.036.
Smith LT, Otterson GA, Plass C: Unraveling the epigenetic code of cancer for therapy. Trends Genet. 2007, 23: 449-456. 10.1016/j.tig.2007.07.005.
Sidransky D: Nucleic acid-based methods for the detection of cancer. Science. 1997, 278: 1054-1059. 10.1126/science.278.5340.1054.
Yu J, Zhu T, Wang Z, Ahang H, Qian Z, Xu H, Gao B, Wang W, Gu , Meng J, Wang J, Feng X, Li Y, Yao X, Zhu J: A Novel set of DNA methylation markers in urine sedments for sensitive/specific detection of bladder cancer. Clin Cancer Res. 2007, 13: 7296-7304. 10.1158/1078-0432.CCR-07-0861.
Dulaimi E, Uzzo RG, Greenberg RE, Al-Saleem T, Cairns P: Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin Cancer Res. 2004, 10: 1887-1893. 10.1158/1078-0432.CCR-03-0127.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/8/238/prepub
Acknowledgements
To the staff at Amaral Carvalho Hospital, Jaú, SP, Brazil for the agreement to contribute to this study, especially to Dr. Renato Costa Prado from the Department of Urology; Dr. Adauto José Ferreira Nunes and Dr. Francisco Carlos Quevedo from the Department of Pathology. To João Paulo de Castro Marcondes, Merielen Garcia Nascimento and Bruna Liboni for other contributions. PDN was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 03/11730-8). This study was financially supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 04/00108-7) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PDN assisted in the study design, in defining the casuistic used, in collecting fresh bladder samples, carrying out the molecular genetic studies and performing the statistical analysis. CAR assisted in the study design, in defining the casuistic used, carrying out the molecular genetic studies and performing the statistical analysis. FPF carried out the molecular genetic studies. JLVC performed the histopathological analysis and classified all fresh bladder samples. MLCSO carried out the cytological analysis of all urinary bladder washings. JG collected urinary bladder washing samples. DMFS was responsible for the study coordination, assisted in the design of the study and in defining the casuistic used. All authors helped to draft the manuscript, and to read and approve the final version.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Negraes, P.D., Favaro, F.P., Camargo, J.L.V. et al. DNA methylation patterns in bladder cancer and washing cell sediments: a perspective for tumor recurrence detection. BMC Cancer 8, 238 (2008). https://doi.org/10.1186/1471-2407-8-238
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-8-238